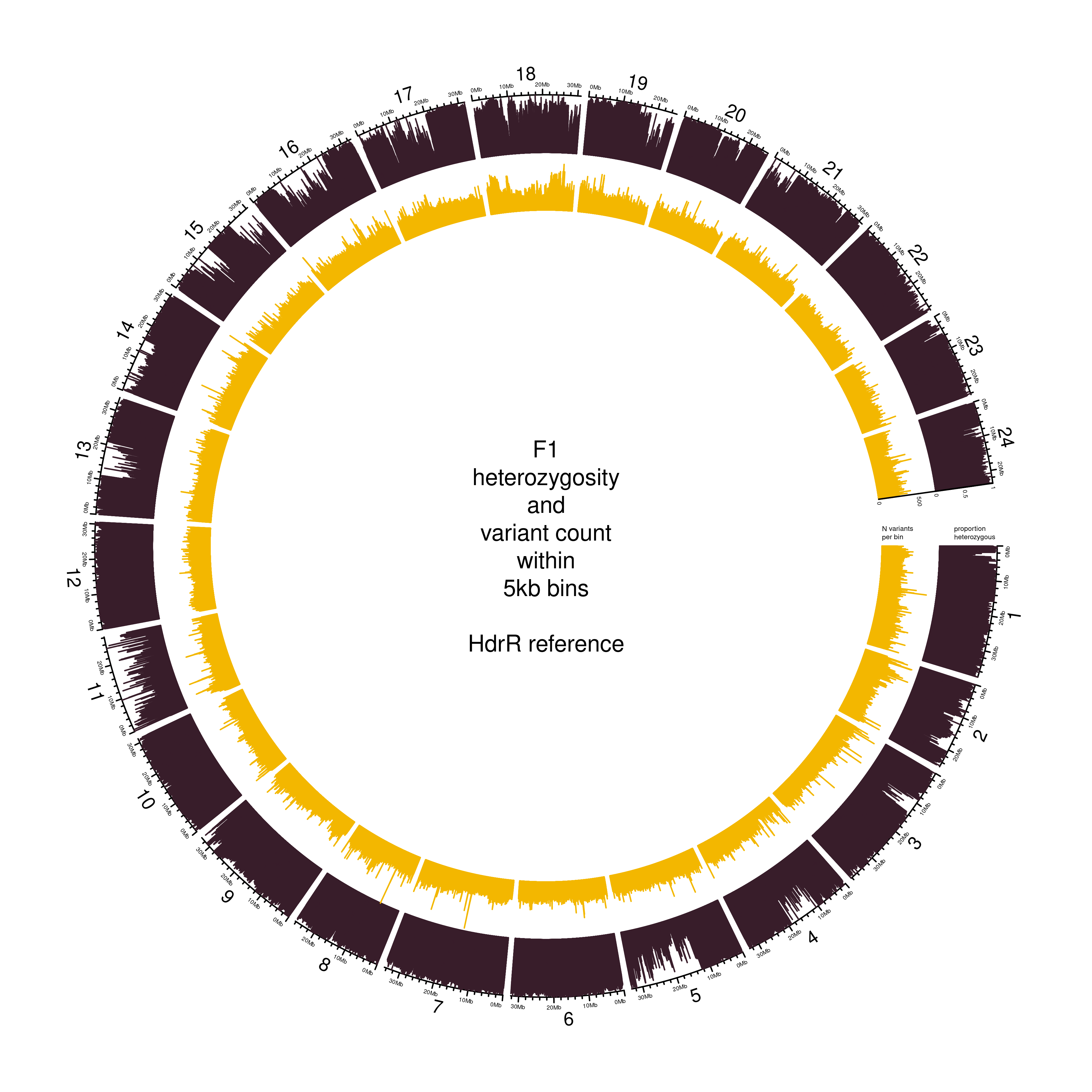
We next examined the level of heterozygosity in the F1 generation from the Cab-Kaga cross. The pipelines used to align and call variants for this sample are set out here and here. Figure 4.1 shows the level of heterozygosity across the genome of the F1 hybrid in brown measured by the proportion of heterozygous SNPs within 5-kb bins (brown), and the number of SNPs in each bin (yellow). Approximately half the chromosomes show inconsistent heterozygosity, with a mean heterozygosity across all bins of 67%. This lower level of apparent heterozygosity than expected was likely caused by the low levels of homozygosity in the Kaga F0 parent.
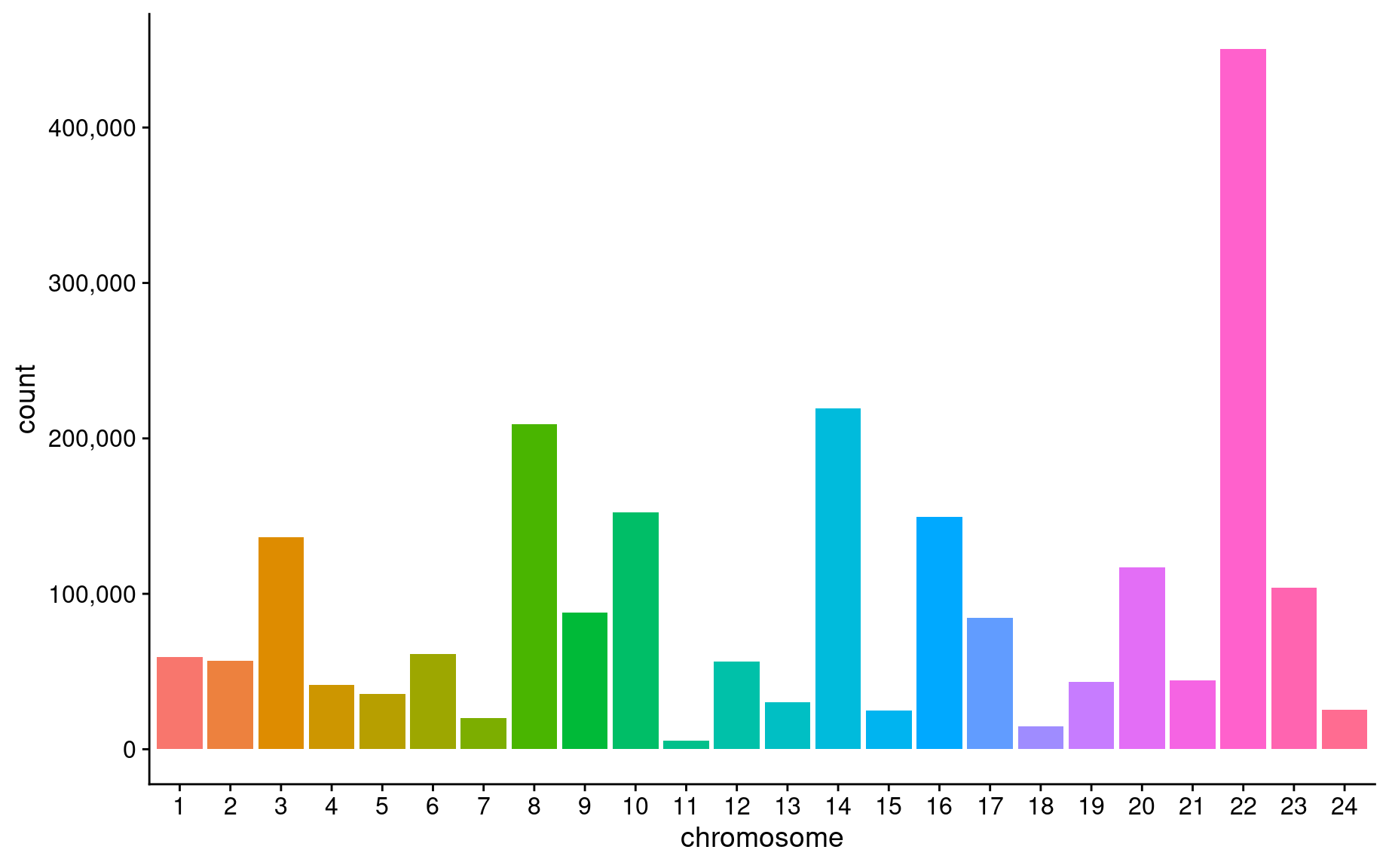
For the purpose of mapping the F2 sample sequences to the genomes of their parental strains, we selected only biallelic SNPs that were homozygous-divergent in the F0 generation (i.e. homozygous reference allele in Cab and homozygous alternative allele in Kaga or vice versa) and heterozygous in the F1 generation. The number of SNPs that met these criteria per chromosome are set out in Figure 4.2. The strong homozygosity of Kaga on chr22 is likely responsible for the much greater number of loci on that chromosome that can be used for calling genoytpes in the F2 generation, and highlights the importance of the parental strains being highly homozygous when used in experimental crosses such as this.