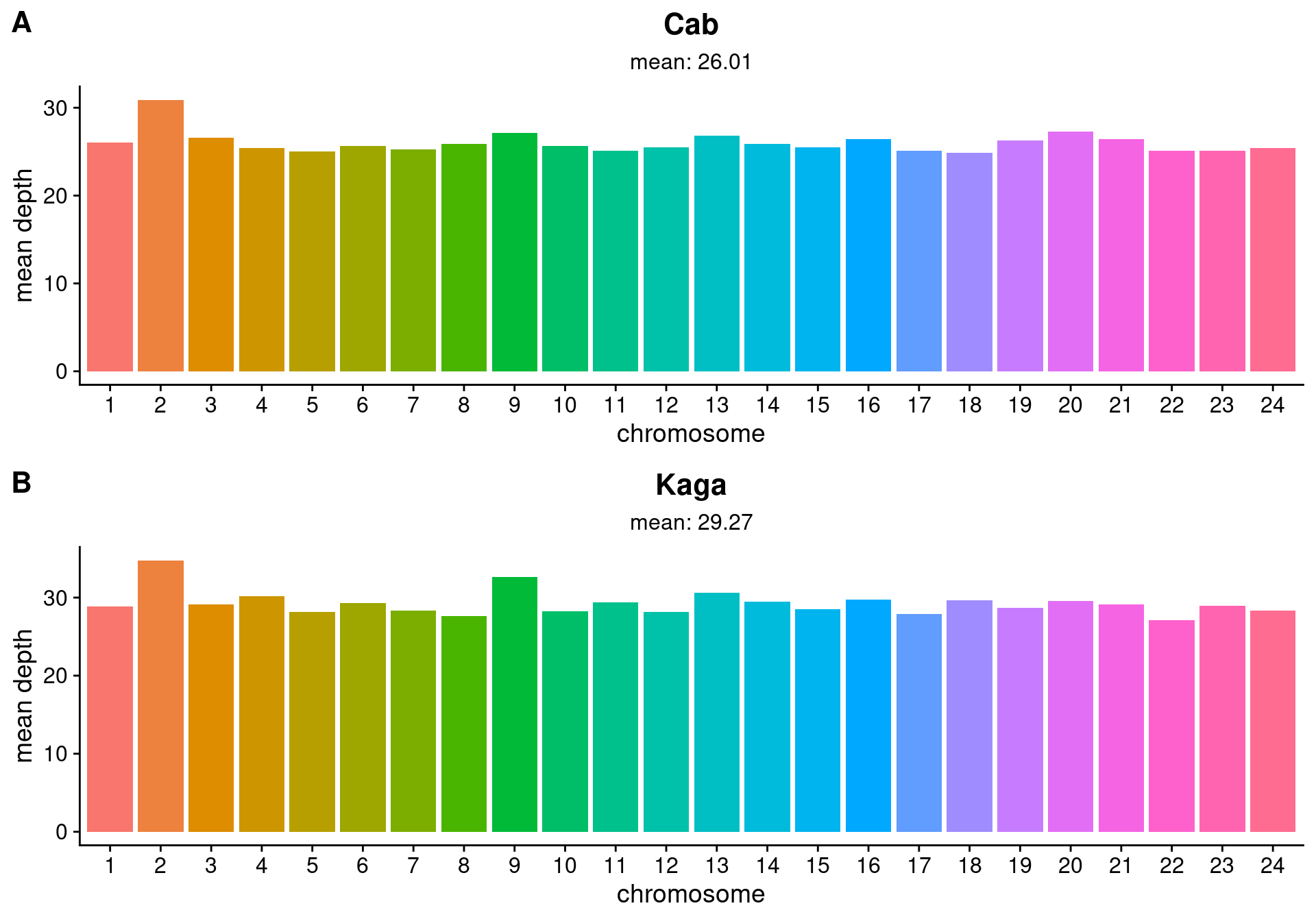
Ali Seleit extracted DNA from the F0, F1, and F2, and sequenced the F0 and F1 samples with the Illumina platform at high coverage (~26x for Cab and ~29x for Kaga), as measured by SAMtools(Danecek et al. 2021). Figure 3.1 sets out the mean sequencing depth within each chromosome and across the whole genome for the Cab and Kaga F0 samples. We then sequenced the F2 samples at low coverage (~1x), which would be sufficient to map their genotypes back to the genotypes of their parental strains (see Chapter 5 for further details).
Before proceeding to map the F2 sequences to the genotypes of the F0 generation, we first investigated the levels of homozygosity in the F0Cab and Kaga strains, as this would affect our ability to accurately call the F2 generation. That is to say, for regions where either F0 parent is consistently heterozygous, it would be difficult to determine the parent from which a particular F2 individual derived its chromosomes at that locus. We therefore aligned the high-coverage sequencing data for the F0Cab and Kaga strains to the medaka HdrR reference (Ensembl release 104, build ASM223467v1) using BWA-MEM2(Vasimuddin et al. 2019), sorted the aligned .sam files, marked duplicate reads, and merged the paired reads with picard (“Picard Toolkit” 2019), and indexed the .bam files with SAMtools(Li et al. 2009). The Snakemake modules used to map and align these samples are set out here and here.
To call variants, we followed the GATK best practices (to the extent they were applicable) (McKenna et al. 2010; DePristo et al. 2011; Van der Auwera and O’Connor 2020) with GATK’s HaplotypeCaller and GenotypeGVCFs tools (Poplin et al. 2018), then merged all calls into a single .vcf file with picard (“Picard Toolkit” 2019). Finally, we extracted the biallelic calls for Cab and Kaga with bcftools (Danecek et al. 2021), counted the number of SNPs within non-overlapping, 5-kb bins, and calculated the proportion of SNPs within each bin that were homozygous.
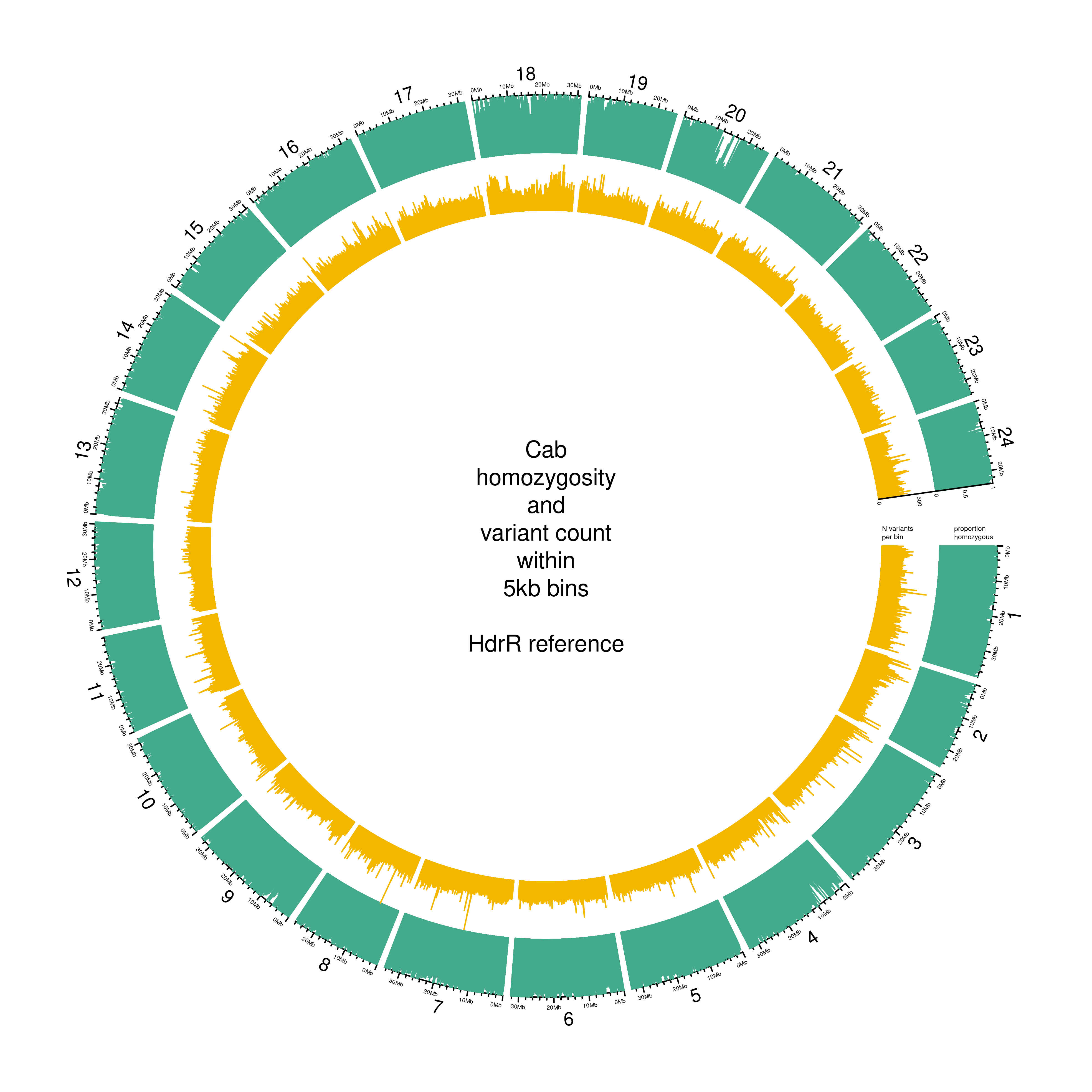
Figure 3.2 is a circos plot generated with circlize (Gu et al. 2014) for the Cab F0 strain used in this experiment, featuring the proportion of homozygous SNPs per 5-kb bin (green), and the total number of SNPs in each bin (yellow). As expected for a strain that has been inbred for over 10 generations, the mean homozygosity for this strain is high, with a mean proportion of homozygosity across all bins of 83%.
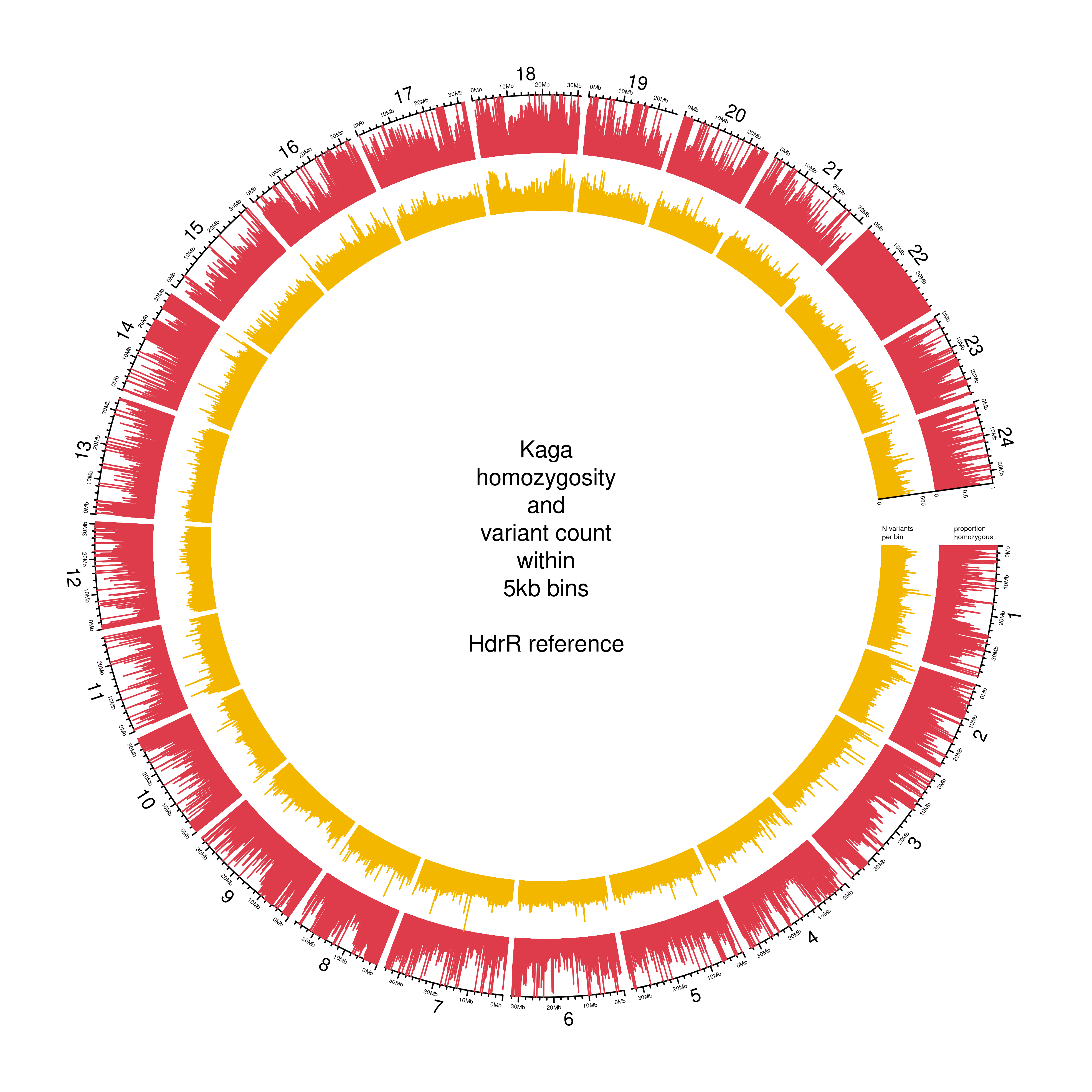
However, the levels of homozygosity in the Kaga strain used in this experiment was far lower, with a mean homozygosity across all bins of only 31% (Figure 3.3). This was a surprise, as it is an established strain that has been extant for decades, and we therefore expected the level of homozygosity to be commensurate with that observed in the Cab strain. An obvious exception is chr22, for which Kaga appears to be homozygous across its entire length.
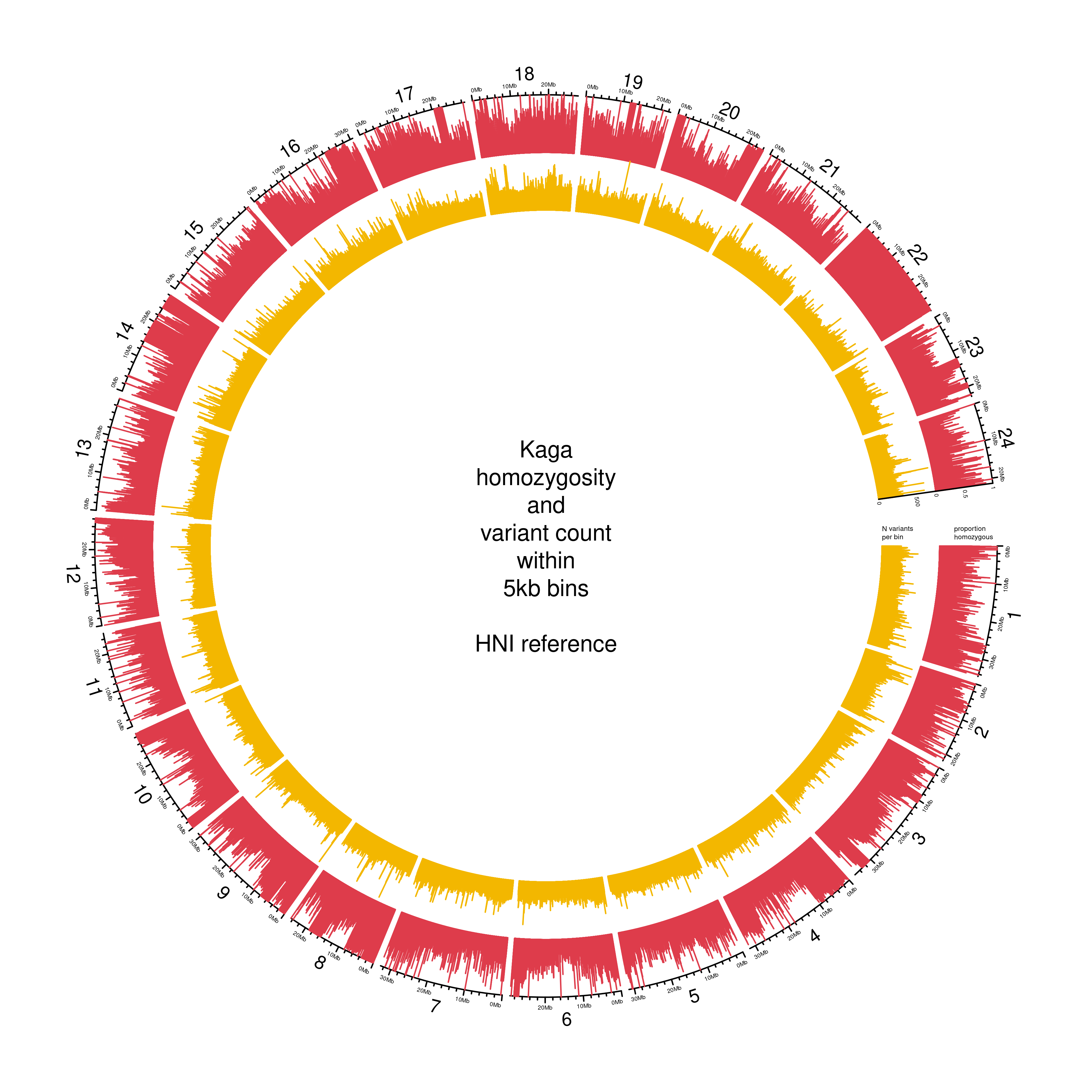
To determine whether the low levels of observed homozygosity in Kaga was affected by its alignments to the southern Japanese HdrR reference, we also aligned the F0 samples to the northern Japanese HNI reference (Figure 3.4). This did not make differences to the levels of observed homozygosity in either sample, which gave us confidence that the low homozygosity observed in Kaga was not driven by reference bias. The low homozygosity of this Kaga individual must have resulted from the strain having been contaminated at some stage by breeding with a different inbred strain prior to when they received the individuals.
Danecek, Petr, James K Bonfield, Jennifer Liddle, John Marshall, Valeriu Ohan, Martin O Pollard, Andrew Whitwham, et al. 2021. “Twelve Years of SAMtools and BCFtools.”GigaScience 10 (2): giab008. https://doi.org/10.1093/gigascience/giab008.
DePristo, Mark A., Eric Banks, Ryan Poplin, Kiran V. Garimella, Jared R. Maguire, Christopher Hartl, Anthony A. Philippakis, et al. 2011. “A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data.”Nature Genetics 43 (5, 5): 491–98. https://doi.org/10.1038/ng.806.
Gu, Zuguang, Lei Gu, Roland Eils, Matthias Schlesner, and Benedikt Brors. 2014. “Circlize Implements and Enhances Circular Visualization in R.”Bioinformatics 30 (19): 2811–12. https://doi.org/10.1093/bioinformatics/btu393.
Li, Heng, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, Richard Durbin, and 1000 Genome Project Data Processing Subgroup. 2009. “The Sequence Alignment/Map (SAM) Format and SAMtools.”Bioinformatics 25 (16): 2078–79.
McKenna, Aaron, Matthew Hanna, Eric Banks, Andrey Sivachenko, Kristian Cibulskis, Andrew Kernytsky, Kiran Garimella, et al. 2010. “The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data.”Genome Research 20 (9): 1297–1303. https://doi.org/10.1101/gr.107524.110.
Poplin, Ryan, Valentin Ruano-Rubio, Mark A. DePristo, Tim J. Fennell, Mauricio O. Carneiro, Geraldine A. Van der Auwera, David E. Kling, et al. 2018. “Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples.”bioRxiv. https://doi.org/10.1101/201178.
Van der Auwera, Geraldine A., and Brian D. O’Connor. 2020. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. O’Reilly Media.
Vasimuddin, Md, Sanchit Misra, Heng Li, and Srinivas Aluru. 2019. “Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems.” In 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS), 314–24. IEEE.