Nucleotide diversity
2022-02-09
1 Load libraries
library(here)
library(tidyverse)
library(karyoploteR)
library(plotly)
library(circlize)
library(GGally)
library(viridis)2 Snakemake pipeline to process VCF and extract data
https://github.com/brettellebi/mikk_genome/tree/master/code/snakemake/nucleotide_diversity
Steps
- Filter MIKK panel VCF for non-sibling lines (N = 63).
- Use
VCFtools’s--window-pito calculate nucleotide divergence in different-sized windows. - Use
bcftoolsto extractMQfromINFOfield.
3 Process data
Data location: /nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence
3.1 Read in data
3.1.1 MIKK
# Set location of data
in_dir = "/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/mikk/all"
in_files = list.files(in_dir, full.names = T)
dat_list = lapply(in_files, function(IN_FILE) {
# Read in
readr::read_delim(IN_FILE,
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
})
names(dat_list) = basename(in_files) %>%
str_remove(".windowed.pi")3.1.2 Wild Kiyosu
# Set location of data
in_dir = "/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/wild_kiyosu"
in_files = list.files(in_dir, full.names = T)
wk_list = lapply(in_files, function(IN_FILE) {
# Read in
readr::read_delim(IN_FILE,
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
})
names(wk_list) = basename(in_files) %>%
str_remove(".windowed.pi")4 Karyoplot
4.1 Set up scaffold
med_chr_lens = read.table(here::here("data",
"Oryzias_latipes.ASM223467v1.dna.toplevel.fa_chr_counts.txt"),
col.names = c("chr", "end"))
# Add start
med_chr_lens$start = 1
# Reorder
med_chr_lens = med_chr_lens %>%
dplyr::select(chr, start, end) %>%
# remove MT
dplyr::filter(chr != "MT") %>%
# convert to numeric
dplyr::mutate(chr = as.integer(chr)) %>%
# order
dplyr::arrange(chr)
# Create custom genome
med_genome = regioneR::toGRanges(med_chr_lens)4.2 Plot
4.2.1 Compare effect of bin width on noise
# Test with ggplot
dat_list$`1000000` %>%
ggplot() +
geom_line(aes(BIN_MID, PI)) +
facet_grid(cols = vars(CHROM))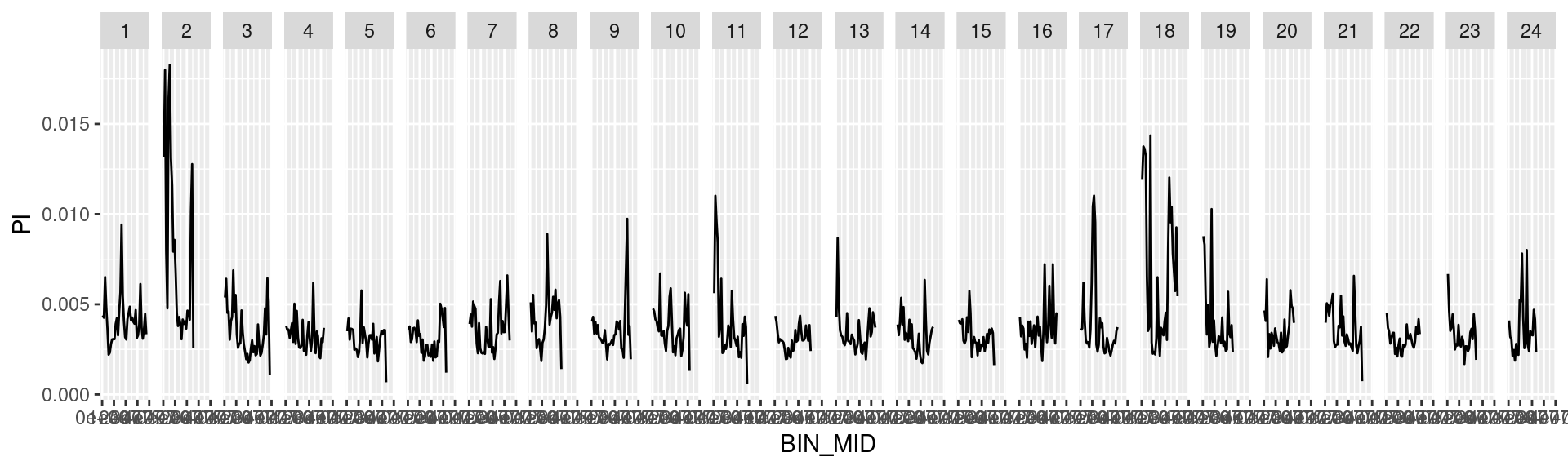
dat_list$`500000` %>%
ggplot() +
geom_line(aes(BIN_MID, PI)) +
facet_grid(cols = vars(CHROM))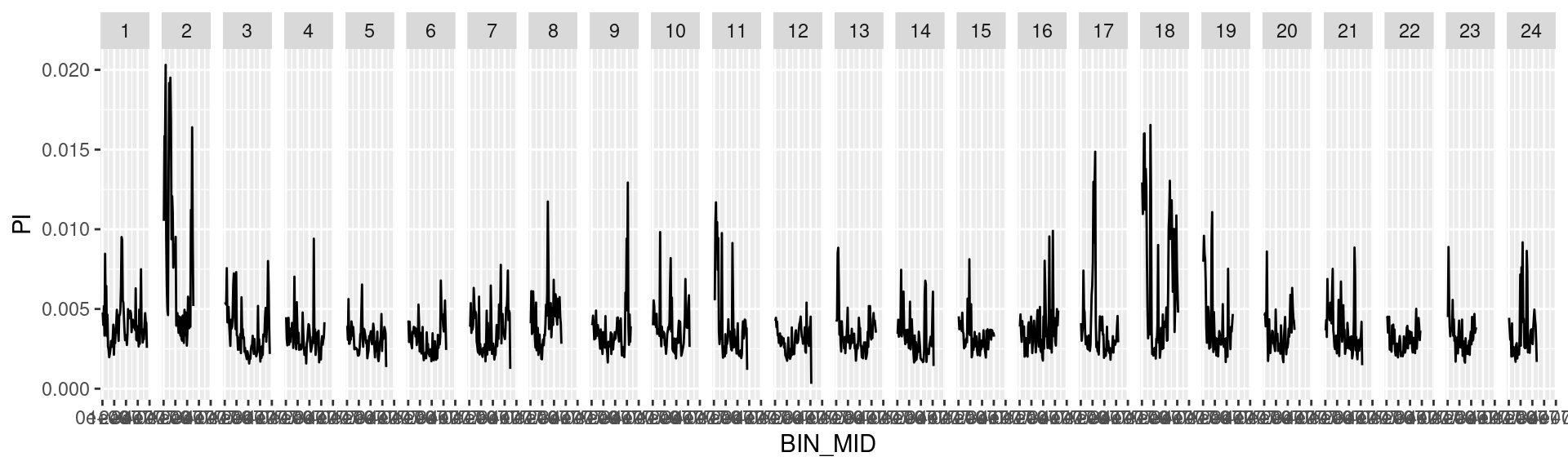
dat_list$`100000` %>%
ggplot() +
geom_line(aes(BIN_MID, PI)) +
facet_grid(cols = vars(CHROM))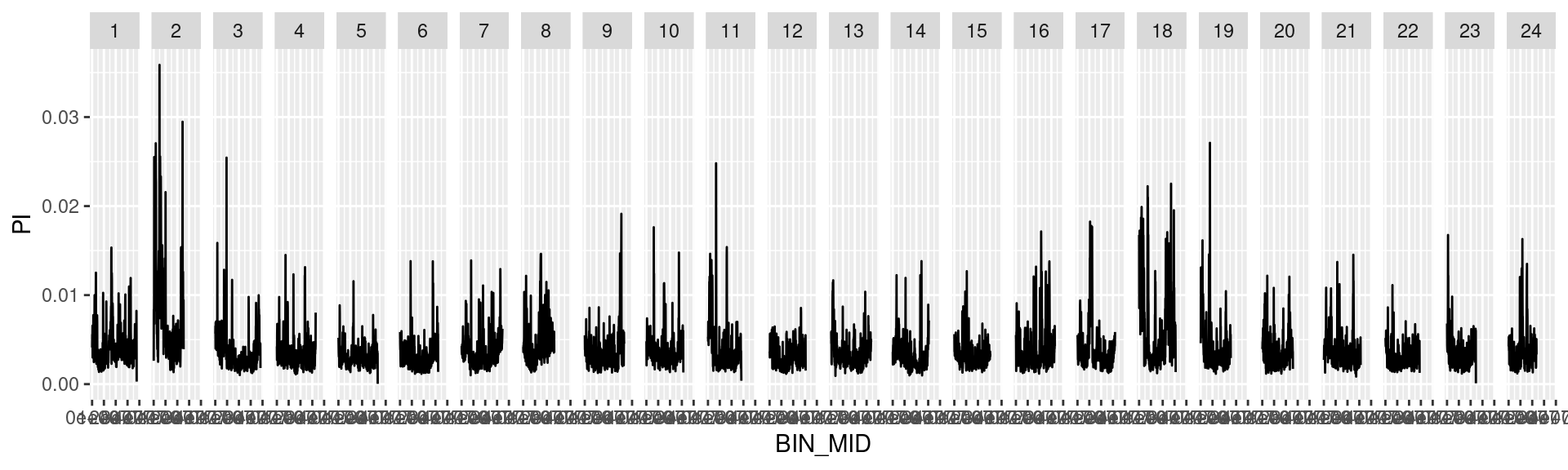
## Get max Y
round.choose <- function(x, roundTo, dir = 1) {
if(dir == 1) { ##ROUND UP
x + (roundTo - x %% roundTo)
} else {
if(dir == 0) { ##ROUND DOWN
x - (x %% roundTo)
}
}
}
y_max = round.choose(max(dat_list$`1000000`$PI), 0.01, 1)# set file name
file_name = paste("20210930_", "1Mb", ".png", sep = "")
file_out = here::here("docs/plots/nucleotide_diversity", file_name)
png(file=file_out,
width=13000,
height=1000,
units = "px",
res = 300)
# Plot ideogram
kp = karyoploteR::plotKaryotype(med_genome, plot.type = 5)
# Add base numbers
karyoploteR::kpAddBaseNumbers(kp, tick.dist = 5000000, cex = 0.7)
# Set y-axis limits
karyoploteR::kpAxis(kp, ymin=0, ymax=y_max )
# Add lines
lwd = 1
karyoploteR::kpLines(kp,
chr = dat_list$`1000000`$CHROM,
x = dat_list$`1000000`$BIN_MID,
y = dat_list$`1000000`$PI,
ymax = y_max,
r0=0, r1 = 1,
lwd = lwd)
dev.off() file_out = here::here("docs/plots/nucleotide_diversity/20210930_1Mb.png")
knitr::include_graphics(file_out)
5 Circos
5.1 Get repeats data
repeats_file = "/nfs/research/birney/users/ian/mikk_genome/repeats/medaka_hdrr_repeats.fixed.gff"
hdrr_reps = readr::read_delim(repeats_file,
delim = "\t",
col_names = F,
skip = 3,
comment = "",
quote = "") %>%
# Remove empty V8 column
dplyr::select(-X8) %>%
# Get class of repeat from third column
dplyr::mutate(class = stringr::str_split(X3, pattern = "#", simplify = T)[, 1]) %>%
# Rename columns
dplyr::rename(chr = X1, tool = X2, class_full = X3, start = X4, end = X5, percent = X6, strand = X7, info = X9)
# Find types of class other than "(GATCCA)n" types
class_types = unique(hdrr_reps$class[grep(")n", hdrr_reps$class, invert = T)])
hdrr_reps = hdrr_reps %>%
# NA for blanks
dplyr::mutate(class = dplyr::na_if(class, "")) %>%
# "misc" for others in "(GATCCA)n" type classes
dplyr::mutate(class = dplyr::if_else(!class %in% class_types, "Miscellaneous", class)) %>%
# rename "Simple_repeat"
dplyr::mutate(class = dplyr::recode(class, "Simple_repeat" = "Simple repeat")) %>%
# filter out NA in `chr` (formerly MT)
dplyr::filter(!is.na(chr))5.2 Calculate proportion of bin covered by repeats
5.2.1 Bin intevals
# Create GRanges object with bins
bin_length = "1000000"
bin_intervals = dat_list[[bin_length]] %>%
dplyr::select(CHROM, BIN_START, BIN_END) %>%
# split into list by chromosome
split(., f = .$CHROM)
# Replace END pos of final bin for each chromsome with actual end pos
bin_intervals = lapply(1:length(bin_intervals), function(CHR){
# Get end position for target chromosome
end_pos = med_chr_lens %>%
dplyr::filter(chr == CHR) %>%
dplyr::pull(end)
# Replace final bin end position
out = bin_intervals[[CHR]]
out$BIN_END[nrow(out)] = end_pos
return(out)
}) %>%
dplyr::bind_rows()
# Convert to data frame
bin_intervals = as.data.frame(bin_intervals)
# Convert to GRanges
bin_ranges = regioneR::toGRanges(bin_intervals)5.2.2 Repeats
# Convert `hdrr_reps` to GRanges
hdrr_ranges = GenomicRanges::makeGRangesFromDataFrame(hdrr_reps,
keep.extra.columns = T,
seqnames.field = "chr",
start.field = "start",
end.field = "end")
# Get non-overlapping regions
hdrr_covered = GenomicRanges::disjoin(hdrr_ranges)5.2.3 Find quantity of repeats overlapping bins
overlaps = GenomicRanges::findOverlaps(bin_ranges, hdrr_covered)
# Split into list by bin
overlaps_list = lapply(unique(overlaps@from), function(BIN){
out = list()
# Get indexes of all repeat ranges overlapping the target BIN
out[["hits"]] = overlaps[overlaps@from == BIN]@to
# Extract those ranges from `hdrr_ranges`
out[["range_hits"]] = hdrr_covered[out[["hits"]]]
# Get number bases covered by each range
out[["widths"]] = out[["range_hits"]] %>%
GenomicRanges::width(.)
# Get summed widths
out[["summed"]] = out[["widths"]] %>%
# Get total bases covered in bin
sum(.)
return(out)
})
overlaps_vec = purrr::map(overlaps_list, function(BIN){
BIN[["summed"]]
}) %>%
unlist(.)
# Add as column to `bin_intervals`
bin_intervals$REPEAT_COV = overlaps_vec
# Caclulate proportion
bin_intervals = bin_intervals %>%
dplyr::mutate(REPEAT_PROP = REPEAT_COV / (BIN_END - BIN_START + 1))5.3 Read in mapping quality scores
in_file = "/nfs/research/birney/users/ian/mikk_genome/mapping_quality/mapping_quality.csv"
mq_df = readr::read_csv(in_file,
col_names = c("CHROM", "POS", "MQ"),
col_types = c("cid")) %>%
# remove 'MT'
dplyr::filter(!CHROM == "MT") %>%
# make CHROM integer
dplyr::mutate(CHROM = as.integer(CHROM))
# Bin and get means
mq_list = mq_df %>%
split(., f = .$CHROM)
# Bin into 1 Mb intervals
binned_mq_list = purrr::map(mq_list, function(CHR){
# Set intervals
intervals = seq(1, max(CHR$POS), by = 1000000)
# add final length
if (max(intervals) != max(CHR$POS)) {
intervals = c(intervals, max(CHR$POS))
}
# bin
CHR = CHR %>%
dplyr::mutate(BIN = cut(POS,
breaks = intervals,
labels = F,
include.lowest = T))
})
# calculate mean MQ within each bin
mean_mq = purrr::map(binned_mq_list, function(CHR){
CHR %>%
# replace Inf with NA
dplyr::mutate(MQ = dplyr::na_if(MQ, Inf)) %>%
# Group by BIN
dplyr::group_by(BIN) %>%
# Calculate mean MQ within each bin
dplyr::summarise(MEAN_MQ = mean(MQ, na.rm = T))
}) %>%
# bind into single data frame
dplyr::bind_rows(.id = "CHR")## Warning: One or more parsing issues, see `problems()` for details5.4 Bind data frames
# Bind data frames
pi_repeat_df = cbind(dat_list$`1000000`,
PI_WK = wk_list[["1000000"]]$PI,
REPEAT_PROP = bin_intervals$REPEAT_PROP,
MEAN_MQ = mean_mq$MEAN_MQ)5.5 Pairs plot
pairs_plot = GGally::ggpairs(pi_repeat_df, columns = c(5, 7, 8, 9),
lower = list(continuous = wrap("points", alpha = 0.2, size = 0.5),
combo = wrap("dot_no_facet", alpha = 0.4)))
plotly::ggplotly(pairs_plot)## Warning: Can only have one: highlight
## Warning: Can only have one: highlight
## Warning: Can only have one: highlight5.6 Circos
5.6.1 1 Mb
5.6.1.1 Rearrange data frames
df_circos_1Mb = pi_repeat_df
# take `BIN_END` from `bin_intervals` with chromosome end number for final bin
df_circos_1Mb$BIN_END = bin_intervals$BIN_END5.6.1.2 Plot
out_plot = here::here("docs/plots/nucleotide_diversity", "20211001_circos_1Mb.png")png(out_plot,
width = 20,
height = 20,
units = "cm",
res = 500)
# Set parameters
## Decrease cell padding from default c(0.02, 1.00, 0.02, 1.00)
circos.par(cell.padding = c(0, 0, 0, 0),
track.margin = c(0, 0),
gap.degree = c(rep(1, nrow(med_chr_lens) - 1), 6))
# Initialize plot
circos.initializeWithIdeogram(med_chr_lens,
plotType = c("axis", "labels"),
major.by = 1e7,
axis.labels.cex = 0.25*par("cex"))
# Add MIKK Pi line
circos.genomicTrack(df_circos_1Mb,
panel.fun = function(region, value, ...){
circos.genomicLines(region,
value[[2]],
col = "#49A379",
area = T,
border = karyoploteR::darker("#49A379"))
},
track.height = 0.1,
bg.border = NA,
ylim = c(0, 0.021))
circos.yaxis(side = "right",
at = c(.01, .02),
labels.cex = 0.25*par("cex"),
tick.length = 2
)
circos.text(0, 0.01,
labels = expression(paste("MIKK (", pi, ")", sep = "")),
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '2'.# Add wild Kiyosu line
circos.genomicTrack(df_circos_1Mb,
panel.fun = function(region, value, ...){
circos.genomicLines(region,
value[[4]],
col = "#7A306C",
area = T,
border = karyoploteR::darker("#8A4F7D"))
},
track.height = 0.1,
bg.border = NA,
ylim = c(0, 0.021))
circos.yaxis(side = "right",
at = c(.01, .02),
labels.cex = 0.25*par("cex"),
tick.length = 2
)
circos.text(0, 0.01,
labels = expression(paste("Wild\nKiyosu (", pi, ")", sep = "")),
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '3'.# Add repeat content
circos.genomicTrack(df_circos_1Mb,
panel.fun = function(region, value, ...) {
circos.genomicLines(region,
value[[5]],
type = "h",
col = "#0C1B33",
cex = 0.05,
baseline = 0)
},
track.height = 0.1,
ylim = c(0,0.5),
bg.border = NA)
circos.yaxis(side = "right",
at = c(0, .5, 1),
labels.cex = 0.25*par("cex"),
tick.length = 5
)
circos.text(0, 0.25,
labels = "Repeat\ncontent",
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '4'.# Add mean mapping quality
circos.genomicTrack(df_circos_1Mb,
panel.fun = function(region, value, ...) {
circos.genomicLines(region,
value[[6]],
type = "h",
col = "#FF6978",
cex = 0.05,
baseline = 20)
},
track.height = 0.1,
ylim = c(20,65),
bg.border = NA)
circos.yaxis(side = "right",
at = c(20, 50),
labels.cex = 0.25*par("cex"),
tick.length = 5
)
circos.text(0, 40,
labels = "Mean MQ",
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '5'.# Add baseline
#circos.xaxis(h = "bottom",
# labels = F,
# major.tick = F)
# Print label in center
text(0, 0, "Nucleotide diversity in\nMIKK and wild Kiyosu medaka,\nrepeat content,\nand mapping quality\nin 1 Mb windows")
circos.clear()
dev.off()## png
## 2knitr::include_graphics(out_plot)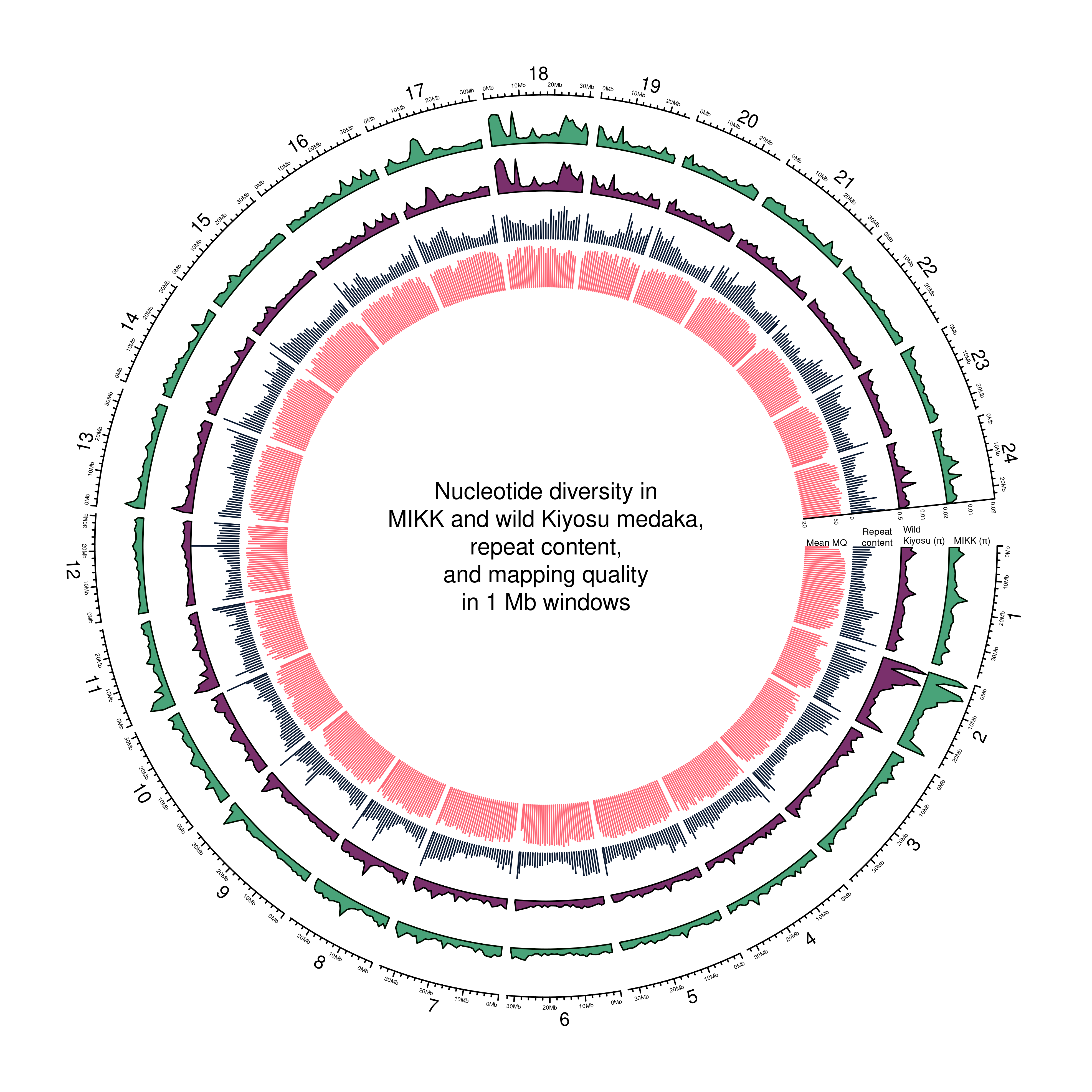
5.6.2 500 Kb
5.6.2.1 Create data frame
5.6.2.1.1 Repeats
# Create GRanges object with bins
bin_length = "500000"
bin_intervals_500 = dat_list[[bin_length]] %>%
dplyr::select(CHROM, BIN_START, BIN_END) %>%
# split into list by chromosome
split(., f = .$CHROM)
# Replace END pos of final bin for each chromsome with actual end pos
bin_intervals_500 = lapply(1:length(bin_intervals_500), function(CHR){
# Get end position for target chromosome
end_pos = med_chr_lens %>%
dplyr::filter(chr == CHR) %>%
dplyr::pull(end)
# Replace final bin end position
out = bin_intervals_500[[CHR]]
out$BIN_END[nrow(out)] = end_pos
return(out)
}) %>%
dplyr::bind_rows()
# Convert to data frame
bin_intervals_500 = as.data.frame(bin_intervals_500)
# Convert to GRanges
bin_ranges = regioneR::toGRanges(bin_intervals_500)
overlaps = GenomicRanges::findOverlaps(bin_ranges, hdrr_covered)
# Split into list by bin
overlaps_list = lapply(unique(overlaps@from), function(BIN){
out = list()
# Get indexes of all repeat ranges overlapping the target BIN
out[["hits"]] = overlaps[overlaps@from == BIN]@to
# Extract those ranges from `hdrr_ranges`
out[["range_hits"]] = hdrr_covered[out[["hits"]]]
# Get number bases covered by each range
out[["widths"]] = out[["range_hits"]] %>%
GenomicRanges::width(.)
# Get summed widths
out[["summed"]] = out[["widths"]] %>%
# Get total bases covered in bin
sum(.)
return(out)
})
overlaps_vec = purrr::map(overlaps_list, function(BIN){
BIN[["summed"]]
}) %>%
unlist(.)
# Add as column to `bin_intervals_500`
bin_intervals_500$REPEAT_COV = overlaps_vec
# Caclulate proportion
bin_intervals_500 = bin_intervals_500 %>%
dplyr::mutate(REPEAT_PROP = REPEAT_COV / (BIN_END - BIN_START + 1))5.6.2.1.2 MQ
# Bin into 1 Mb intervals
binned_mq_list_500 = purrr::map(mq_list, function(CHR){
# Set intervals
intervals = seq(1, max(CHR$POS), by = 500000)
# add final length
if (max(intervals) != max(CHR$POS)) {
intervals = c(intervals, max(CHR$POS))
}
# bin
CHR = CHR %>%
dplyr::mutate(BIN = cut(POS,
breaks = intervals,
labels = F,
include.lowest = T))
})
# calculate mean MQ within each bin
mean_mq_500 = purrr::map(binned_mq_list_500, function(CHR){
CHR %>%
# replace Inf with NA
dplyr::mutate(MQ = dplyr::na_if(MQ, Inf)) %>%
# Group by BIN
dplyr::group_by(BIN) %>%
# Calculate mean MQ within each bin
dplyr::summarise(MEAN_MQ = mean(MQ, na.rm = T))
}) %>%
# bind into single data frame
dplyr::bind_rows(.id = "CHR")5.6.2.1.3 Final
df_circos_500kb = cbind(dat_list$`500000`,
PI_WK = wk_list[["500000"]]$PI,
REPEAT_PROP = bin_intervals_500$REPEAT_PROP,
MEAN_MQ = mean_mq_500$MEAN_MQ)
# Add bin ends at the end of each chromosome to match real chromosome ends
df_circos_500kb$BIN_END = bin_intervals_500$BIN_ENDout_plot = here::here("docs/plots/nucleotide_diversity/20211001_circos_500kb.png")png(out_plot,
width = 20,
height = 20,
units = "cm",
res = 500)
# Set parameters
## Decrease cell padding from default c(0.02, 1.00, 0.02, 1.00)
circos.par(cell.padding = c(0, 0, 0, 0),
track.margin = c(0, 0),
gap.degree = c(rep(1, nrow(med_chr_lens) - 1), 6))
# Initialize plot
circos.initializeWithIdeogram(med_chr_lens,
plotType = c("axis", "labels"),
major.by = 1e7,
axis.labels.cex = 0.25*par("cex"))
# Add MIKK Pi line
circos.genomicTrack(df_circos_500kb,
panel.fun = function(region, value, ...){
circos.genomicLines(region,
value[[2]],
col = "#49A379",
area = T,
border = karyoploteR::darker("#49A379"))
},
track.height = 0.1,
bg.border = NA,
ylim = c(0, 0.023))
circos.yaxis(side = "right",
at = c(.01, .02),
labels.cex = 0.25*par("cex"),
tick.length = 2
)
circos.text(0, 0.01,
labels = expression(paste("MIKK (", pi, ")", sep = "")),
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '2'.# Add wild Kiyosu line
circos.genomicTrack(df_circos_500kb,
panel.fun = function(region, value, ...){
circos.genomicLines(region,
value[[4]],
col = "#7A306C",
area = T,
border = karyoploteR::darker("#8A4F7D"))
},
track.height = 0.1,
bg.border = NA,
ylim = c(0, 0.023))
circos.yaxis(side = "right",
at = c(.01, .02),
labels.cex = 0.25*par("cex"),
tick.length = 2
)
circos.text(0, 0.01,
labels = expression(paste("Wild\nKiyosu (", pi, ")", sep = "")),
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '3'.# Add repeat content
circos.genomicTrack(df_circos_500kb,
panel.fun = function(region, value, ...) {
circos.genomicLines(region,
value[[5]],
type = "h",
col = "#0C1B33",
lwd = 0.4,
cex = 0.05,
baseline = 0)
},
track.height = 0.1,
ylim = c(0,0.65),
bg.border = NA)
circos.yaxis(side = "right",
at = c(0, .5, 1),
labels.cex = 0.25*par("cex"),
tick.length = 5
)
circos.text(0, 0.325,
labels = "Repeat\ncontent",
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '4'.# Add mean mapping quality
circos.genomicTrack(df_circos_500kb,
panel.fun = function(region, value, ...) {
circos.genomicLines(region,
value[[6]],
type = "h",
col = "#FF6978",
lwd = 0.4,
cex = 0.05,
baseline = 20)
},
track.height = 0.07,
ylim = c(20,65),
bg.border = NA)
circos.yaxis(side = "right",
at = c(20, 50),
labels.cex = 0.25*par("cex"),
tick.length = 5
)
circos.text(0, 40,
labels = "Mean MQ",
sector.index = "1",
facing = "clockwise",
adj = c(.5, 0),
cex = 0.4*par("cex")) ## Note: 1 point is out of plotting region in sector '1', track '5'.# Print label in center
text(0, 0, "Nucleotide diversity in\nMIKK and wild Kiyosu medaka,\nwith repeat content\nand mapping quality\nin 500 kb windows")
circos.clear()
dev.off()## png
## 2knitr::include_graphics(out_plot)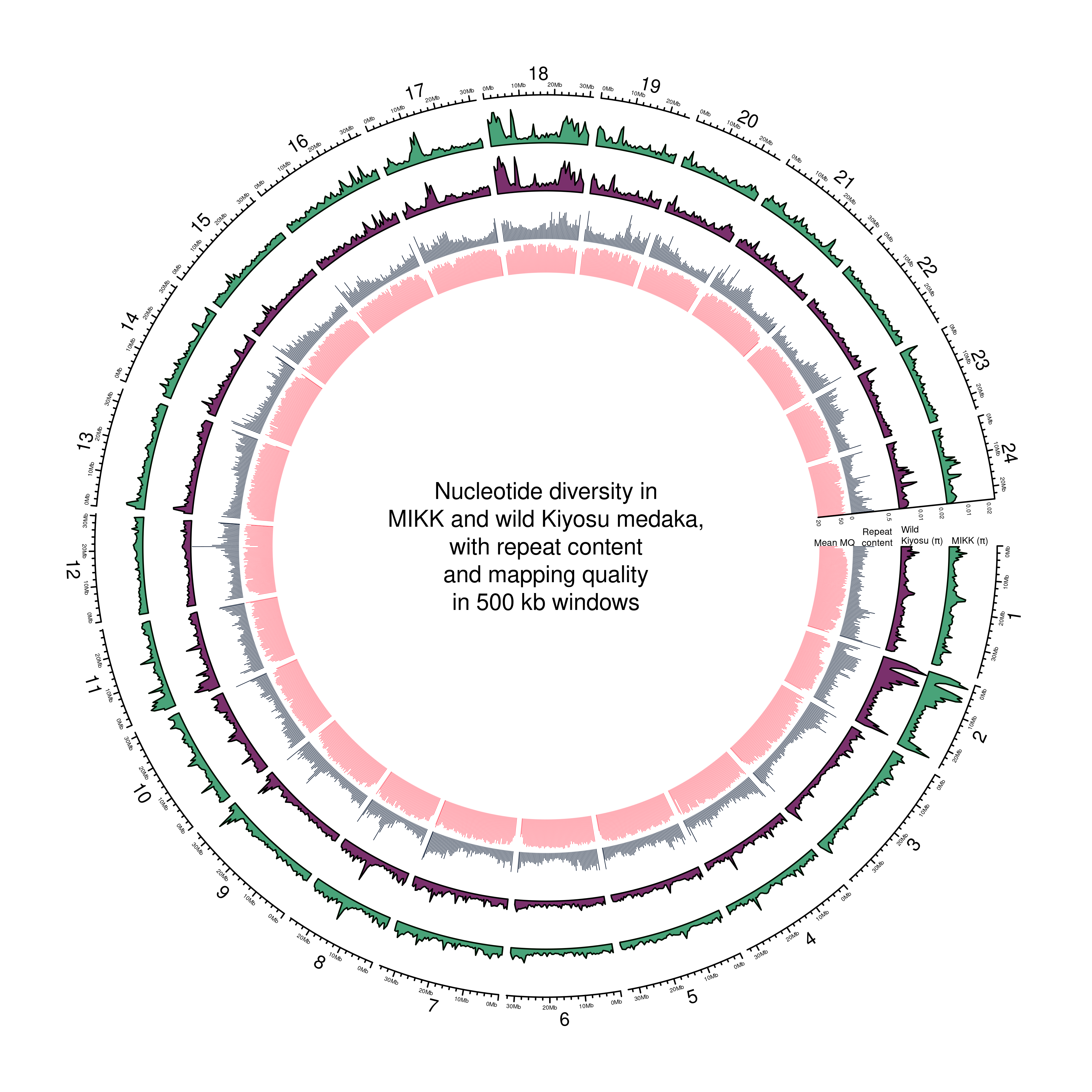
5.6.3 Mean and median Pi
5.6.3.1 63 MIKK samples v 7 wild Kiyosu
# MIKK
mean(df_circos_500kb$PI)## [1] 0.003829434median(df_circos_500kb$PI)## [1] 0.00329243# Wild median Pi
mean(df_circos_500kb$PI_WK)## [1] 0.003685076median(df_circos_500kb$PI_WK)## [1] 0.0031028055.6.3.2 7 MIKK samples v 7 wild Kiyosu
# Read in data
mikk_7 = readr::read_delim("/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/mikk/random/500.windowed.pi",
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
# Mean
mean(mikk_7$PI)## [1] 0.003748531median(mikk_7$PI)## [1] 0.0031976756 Per-individual
6.1 Read in data
6.1.1 MIKK
# Set location of data
in_dir = "/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/mikk/per_sample"
in_files = list.files(in_dir, full.names = T)
mikk_list = lapply(in_files, function(IN_FILE) {
# Read in
readr::read_delim(IN_FILE,
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
})
names(mikk_list) = basename(in_files) %>%
str_remove(".windowed.pi")
# reorder
ordered_lines = tibble::tibble("LINE" = names(mikk_list)) %>%
tidyr::separate(col = LINE, into = c("LINE", "SIB", "EXTRA"), sep = "_") %>%
dplyr::mutate(LINE = as.integer(LINE)) %>%
dplyr::arrange(LINE) %>%
tidyr::unite(col = "ORIGINAL", sep = "_", na.rm = T) %>%
dplyr::pull()## Warning: Expected 3 pieces. Missing pieces filled with `NA` in 62 rows [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16,
## 17, 18, 19, 20, 21, ...].mikk_list = mikk_list[order(match(names(mikk_list), ordered_lines))]
# Bind rows
mikk_df = mikk_list %>%
dplyr::bind_rows(.id = "LINE") %>%
# factor LINE to order
dplyr::mutate(LINE = factor(LINE, levels = names(mikk_list)))6.1.2 Wild
# Set location of data
in_dir = "/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/wild/per_sample"
in_files = list.files(in_dir, full.names = T)
wild_list = lapply(in_files, function(IN_FILE) {
# Read in
readr::read_delim(IN_FILE,
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
})
names(wild_list) = basename(in_files) %>%
str_remove(".windowed.pi")
# Bind rows
wild_df = wild_list %>%
dplyr::bind_rows(.id = "LINE")6.1.3 Combine
pi_df = list("MIKK" = mikk_df,
"WILD" = wild_df) %>%
dplyr::bind_rows(.id = "POPULATION")6.1.4 Boxplot
# Create palette
pal = c("#49A379", "#8A4F7D")
names(pal) = c("MIKK", "WILD")
mikk_wild_boxplot_no_filter = pi_df %>%
ggplot() +
geom_violin(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION)) +
geom_boxplot(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION), width = 0.1) +
theme_bw() +
scale_fill_manual(name = "Population", values = pal) +
scale_colour_manual(name = "Population", values = darker(pal, 80)) +
ylab(expression(paste(log[10], "(", pi, ")", sep = ""))) +
ggtitle("Nucleotide diversity in 500 kb bins (MAPQ >= 50)")
mikk_wild_boxplot_no_filter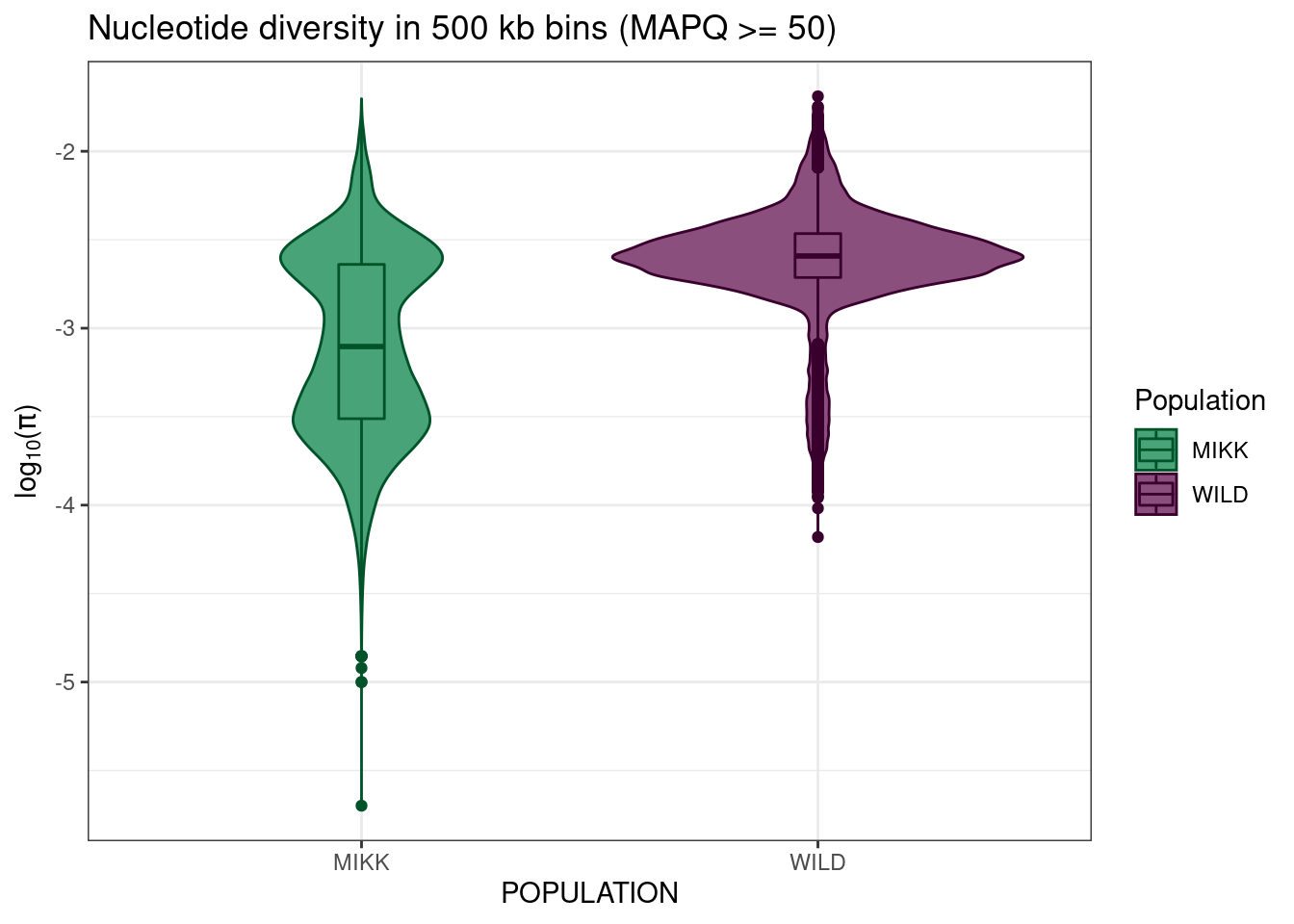
ggsave(here::here("docs/plots/nucleotide_diversity/20211006_mikk_wild_boxplot_no_filter.png"),
plot = mikk_wild_boxplot_no_filter,
device = "png",
width = 8,
height = 6,
units = "in",
dpi = 400)6.1.5 All chromosomes
purrr::map(1:24, function(CHR){
# Create plot
plot_df = mikk_df %>%
# Filter for target CHROM
dplyr::filter(CHROM == CHR) %>%
# Create Mb column
dplyr::mutate(Mb = (BIN_START - 1) / 1e6)
out_plot = plot_df %>%
ggplot() +
geom_col(aes(Mb, PI, fill = PI)) +
facet_grid(rows = vars(LINE), cols = vars(CHROM)) +
theme_bw() +
theme(text = element_text(size = 5)) +
ylim(c(0, max(mikk_df$PI))) +
xlab("500 kb window start position (Mb)") +
ylab(expression(paste("Nucleotide diversity (", pi, ")", sep = ""))) +
ggtitle(paste("Chromosome ", CHR, sep = "")) +
scale_fill_viridis(name = expression(pi), limits = c(0,max(mikk_df$PI)))
# Adjust width dimensions of plot based on length of chromosome
w_dim = mikk_df %>%
dplyr::filter(CHROM == CHR) %>%
dplyr::mutate(Mb = (BIN_START - 1) / 1e6) %>%
dplyr::pull(Mb) %>%
max(.)
w_dim = w_dim * 0.26
# Save
ggsave(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr", paste(CHR, ".png", sep = "")),
plot = out_plot,
device = "png",
width = w_dim,
height = 20,
units = "in",
dpi = 400)
})knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr/1.png"))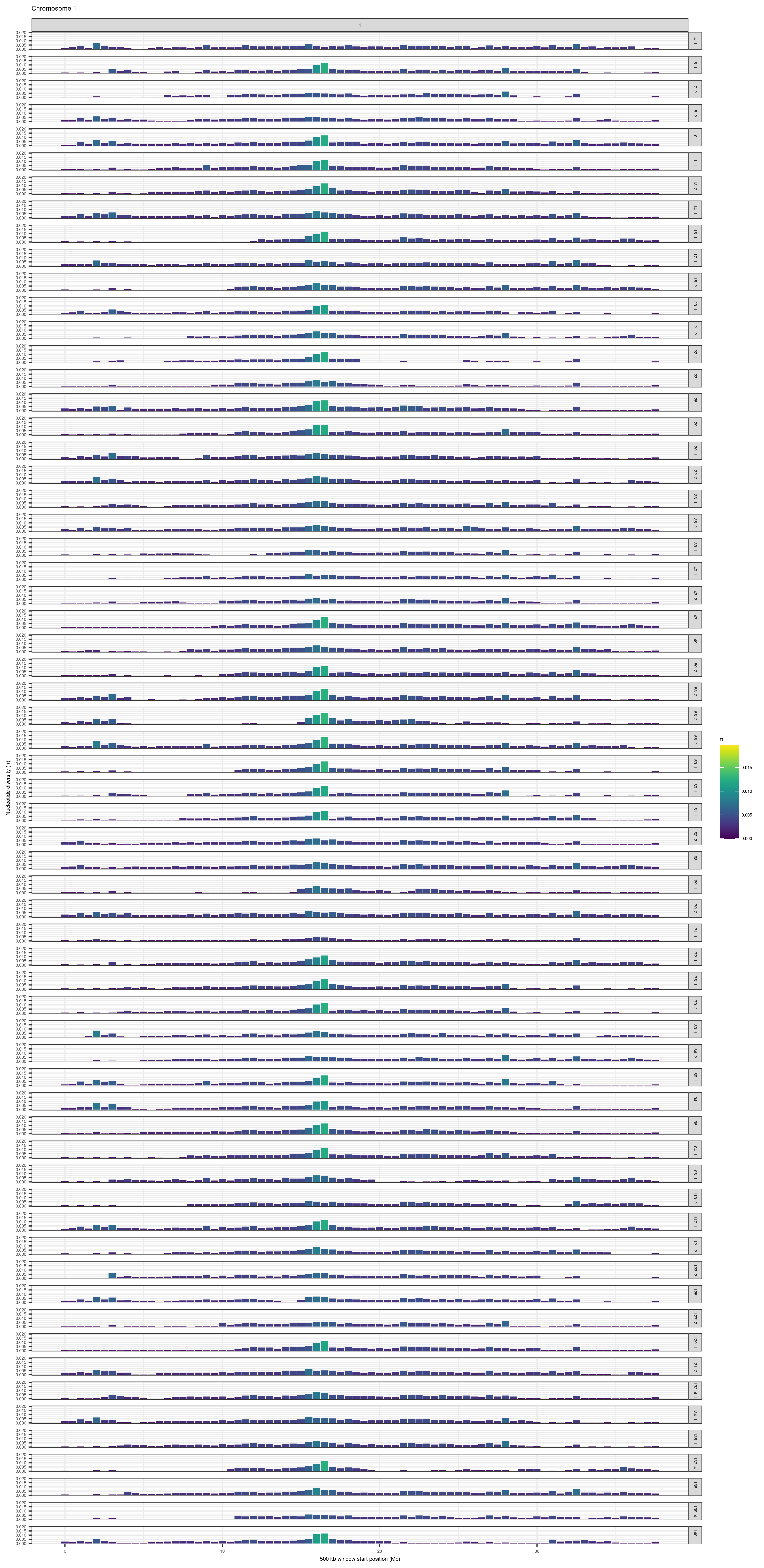
knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr/2.png"))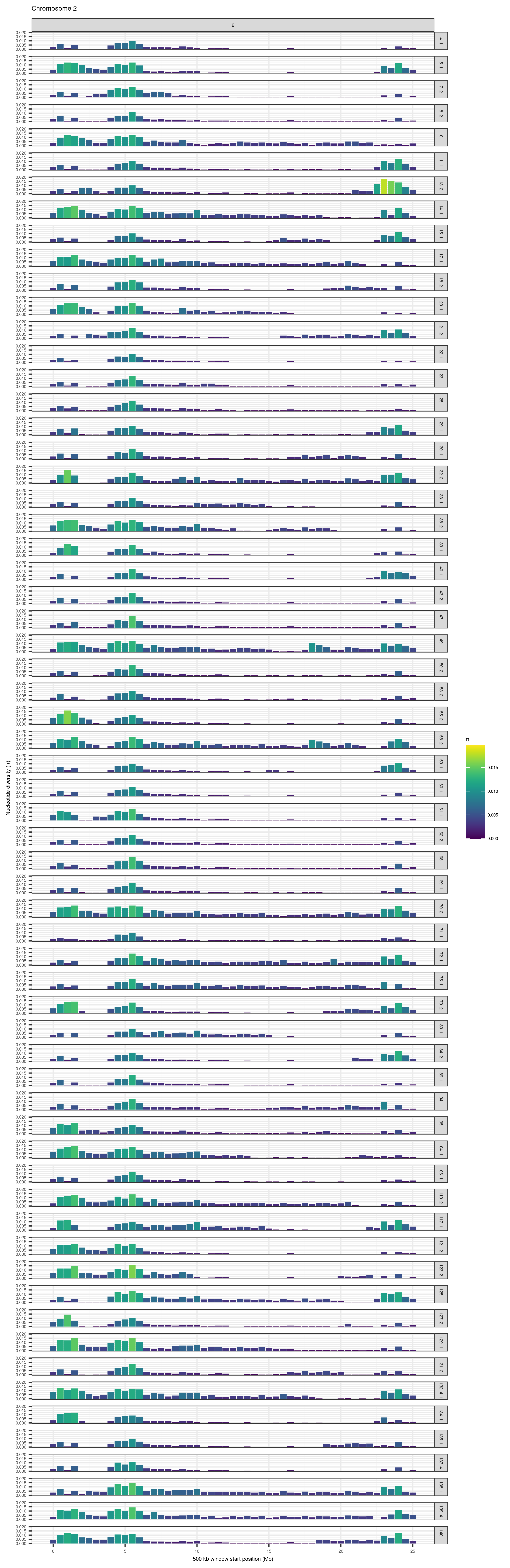
knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr/22.png"))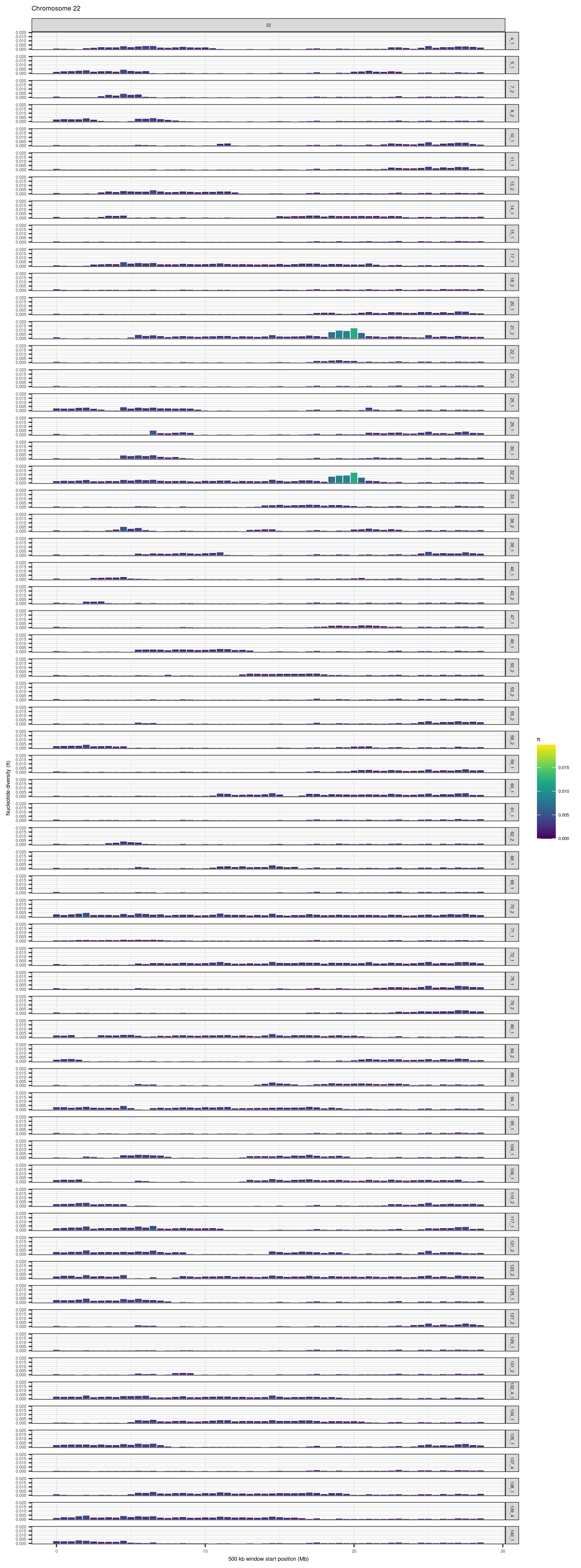
6.1.6 chr1 and 22 for supplementary figures
target_chrs = c(1, 22)
# Create plot
final_plot_df = mikk_df %>%
# Filter for target CHROM
dplyr::filter(CHROM %in% target_chrs) %>%
# Create Mb column
dplyr::mutate(Mb = (BIN_START - 1) / 1e6)
out_plot = final_plot_df %>%
ggplot() +
geom_col(aes(Mb, PI, fill = PI)) +
facet_grid(rows = vars(LINE), cols = vars(CHROM)) +
theme_bw() +
theme(text = element_text(size = 5)) +
ylim(c(0, max(mikk_df$PI))) +
xlab("500 kb window start position (Mb)") +
ylab(expression(paste("Nucleotide diversity (", pi, ")", sep = ""))) +
scale_fill_viridis(name = expression(pi), limits = c(0,max(mikk_df$PI)))
out_plot
# Save
ggsave(here::here("docs/plots/nucleotide_diversity/20211013_chrs_1_22.png"),
plot = out_plot,
device = "png",
width = 15,
height = 20,
units = "in",
dpi = 400)knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr/1.png"))
knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr/2.png"))
knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr/22.png"))
6.2 Investigate QC stats for sample MIKK10_1 to determine appropriate cutoffs
qc_file = "/hps/nobackup/birney/users/ian/mikk_genome/qc_stats/10_1.csv"
qc_df = readr::read_csv(qc_file,
col_names = c("CHROM", "POS", "DP", "MQ", "QD", "GQ"),
col_types = c("ciiddi")) %>%
# Filter for non-missing GQ
dplyr::filter(complete.cases(GQ)) %>%
# Filter out MT
dplyr::filter(CHROM != "MT") %>%
# Re-class CHROM
dplyr::mutate(CHROM = factor(CHROM, levels = 1:24))## Warning: One or more parsing issues, see `problems()` for details6.2.1 Plot
qc_pairs = qc_df %>%
# take sample to prevent over-plotting
dplyr::slice_sample(n = 1e5) %>%
# create log10 for `DP`
dplyr::mutate(LOG_10_DP = log10(DP)) %>%
GGally::ggpairs(.,
columns = c(7, 4, 5, 6),
lower = list(continuous = wrap("points", alpha = 0.2, size = 0.5),
combo = wrap("dot_no_facet", alpha = 0.4)))
qc_pairs## plot: [1,1] [=====>----------------------------------------------------------------------------------------] 6% est: 0s
## plot: [1,2] [===========>----------------------------------------------------------------------------------] 12% est: 2s## Warning in ggally_statistic(data = data, mapping = mapping, na.rm = na.rm, : Removing 1 row that contained a missing value## plot: [1,3] [=================>----------------------------------------------------------------------------] 19% est: 2s## Warning in ggally_statistic(data = data, mapping = mapping, na.rm = na.rm, : Removed 2 rows containing missing values## plot: [1,4] [=======================>----------------------------------------------------------------------] 25% est: 3s
## plot: [2,1] [============================>-----------------------------------------------------------------] 31% est: 2s## Warning: Removed 1 rows containing missing values (geom_point).## plot: [2,2] [==================================>-----------------------------------------------------------] 38% est: 2s## Warning: Removed 1 rows containing non-finite values (stat_density).## plot: [2,3] [========================================>-----------------------------------------------------] 44% est: 2s## Warning in ggally_statistic(data = data, mapping = mapping, na.rm = na.rm, : Removed 2 rows containing missing values## plot: [2,4] [==============================================>-----------------------------------------------] 50% est: 2s## Warning in ggally_statistic(data = data, mapping = mapping, na.rm = na.rm, : Removing 1 row that contained a missing value## plot: [3,1] [====================================================>-----------------------------------------] 56% est: 1s## Warning: Removed 2 rows containing missing values (geom_point).## plot: [3,2] [==========================================================>-----------------------------------] 62% est: 1s## Warning: Removed 2 rows containing missing values (geom_point).## plot: [3,3] [================================================================>-----------------------------] 69% est: 1s## Warning: Removed 2 rows containing non-finite values (stat_density).## plot: [3,4] [=====================================================================>------------------------] 75% est: 1s## Warning in ggally_statistic(data = data, mapping = mapping, na.rm = na.rm, : Removed 2 rows containing missing values## plot: [4,1] [===========================================================================>------------------] 81% est: 1s
## plot: [4,2] [=================================================================================>------------] 88% est: 0s## Warning: Removed 1 rows containing missing values (geom_point).## plot: [4,3] [=======================================================================================>------] 94% est: 0s## Warning: Removed 2 rows containing missing values (geom_point).## plot: [4,4] [==============================================================================================]100% est: 0s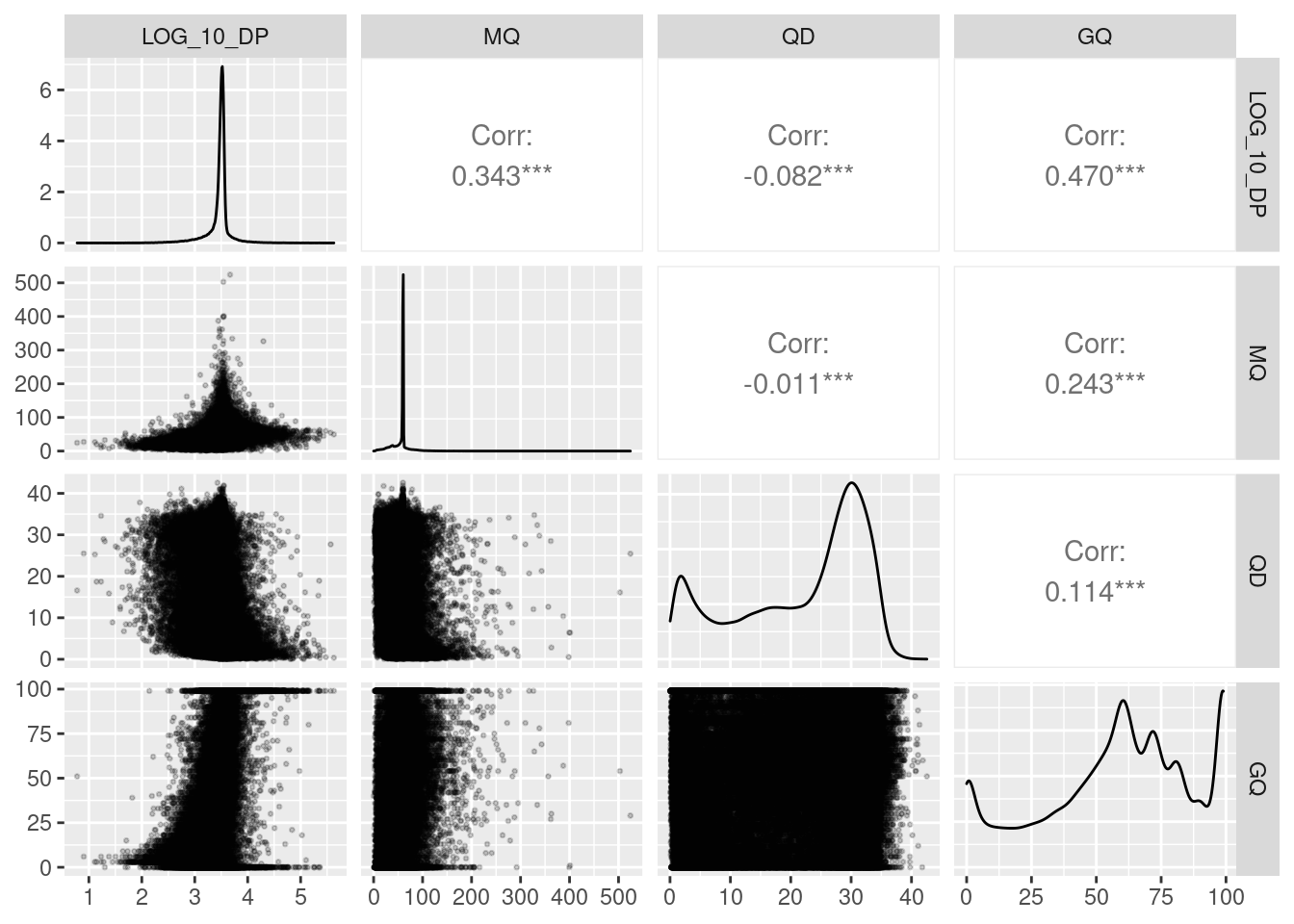
ggsave(here::here("docs/plots/nucleotide_diversity/20211006_qc_pairs.png"),
plot = qc_pairs,
device = "png",
width = 10,
height = 6,
units = "in",
dpi = 400)How many calls have DP < x?
# Total variants
nrow(qc_df)## [1] 27485882# Number of variants with DP < x
length(which(qc_df$DP < 10))## [1] 151length(which(qc_df$DP < 20))## [1] 723length(which(qc_df$DP < 30))## [1] 1823length(which(qc_df$DP < 40))## [1] 3378So few!
Try GQ >= 40 & DP >= 40 & MQ >= 50.
6.3 Read in nucleotide diversity for filtered variants
6.3.1 MIKK
# Set location of data
in_dir = "/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/mikk/filtered"
in_files_mikk_filt = list.files(in_dir, full.names = T)
mikk_list_filt = lapply(in_files_mikk_filt, function(IN_FILE) {
# Read in
readr::read_delim(IN_FILE,
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
})
names(mikk_list_filt) = basename(in_files_mikk_filt) %>%
str_remove(".windowed.pi")
# reorder
ordered_lines = tibble::tibble("LINE" = names(mikk_list_filt)) %>%
tidyr::separate(col = LINE, into = c("LINE", "SIB", "EXTRA"), sep = "_") %>%
dplyr::mutate(LINE = as.integer(LINE)) %>%
dplyr::arrange(LINE) %>%
tidyr::unite(col = "ORIGINAL", sep = "_", na.rm = T) %>%
dplyr::pull()## Warning: Expected 3 pieces. Missing pieces filled with `NA` in 62 rows [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16,
## 17, 18, 19, 20, 21, ...].mikk_list_filt = mikk_list_filt[order(match(names(mikk_list_filt), ordered_lines))]
# Bind rows
mikk_df_filt = mikk_list_filt %>%
dplyr::bind_rows(.id = "LINE") %>%
# factor LINE to order
dplyr::mutate(LINE = factor(LINE, levels = names(mikk_list_filt)))6.3.2 Wild
# Set location of data
in_dir = "/nfs/research/birney/users/ian/mikk_genome/nucleotide_divergence/wild/filtered"
in_files_wild_filt = list.files(in_dir, full.names = T)
wild_list_filt = lapply(in_files_wild_filt, function(IN_FILE) {
# Read in
readr::read_delim(IN_FILE,
delim = "\t",
col_types = c("ciiid")) %>%
# Remove MT
dplyr::filter(CHROM != "MT") %>%
# Make CHR an integer
dplyr::mutate(CHROM = as.integer(CHROM)) %>%
# Create middle of bin
dplyr::mutate(BIN_MID = ((BIN_END - BIN_START - 1) / 2) + BIN_START )
})
names(wild_list_filt) = basename(in_files_wild_filt) %>%
str_remove(".windowed.pi")
# Bind rows
wild_df_filt = wild_list_filt %>%
dplyr::bind_rows(.id = "LINE")6.3.3 Combine
pi_df_filt = list("MIKK" = mikk_df_filt,
"WILD" = wild_df_filt) %>%
dplyr::bind_rows(.id = "POPULATION")6.3.4 All chromosomes
purrr::map(1:24, function(CHR){
# Create plot
plot_df = mikk_df_filt %>%
# Filter for target CHROM
dplyr::filter(CHROM == CHR) %>%
# Create Mb column
dplyr::mutate(Mb = (BIN_START - 1) / 1e6)
out_plot = plot_df %>%
ggplot() +
geom_col(aes(Mb, PI, fill = PI)) +
facet_grid(rows = vars(LINE), cols = vars(CHROM)) +
theme_bw() +
theme(text = element_text(size = 5)) +
ylim(c(0, max(mikk_df$PI))) +
xlab("500 kb window start position (Mb)") +
ylab(expression(paste("Nucleotide diversity (", pi, ")", sep = ""))) +
ggtitle(paste("Chromosome ", CHR, sep = "")) +
scale_fill_viridis(name = expression(pi), limits = c(0,max(mikk_df$PI)))
# Adjust width dimensions of plot based on length of chromosome
w_dim = mikk_df_filt %>%
dplyr::filter(CHROM == CHR) %>%
dplyr::mutate(Mb = (BIN_START - 1) / 1e6) %>%
dplyr::pull(Mb) %>%
max(.)
w_dim = w_dim * 0.26
# Save
ggsave(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr_filtered", paste(CHR, ".png", sep = "")),
plot = out_plot,
device = "png",
width = w_dim,
height = 20,
units = "in",
dpi = 400)
})knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr_filtered/1.png"))
knitr::include_graphics(here::here("docs/plots/nucleotide_diversity/20211006_pi_per_chr_filtered/22.png"))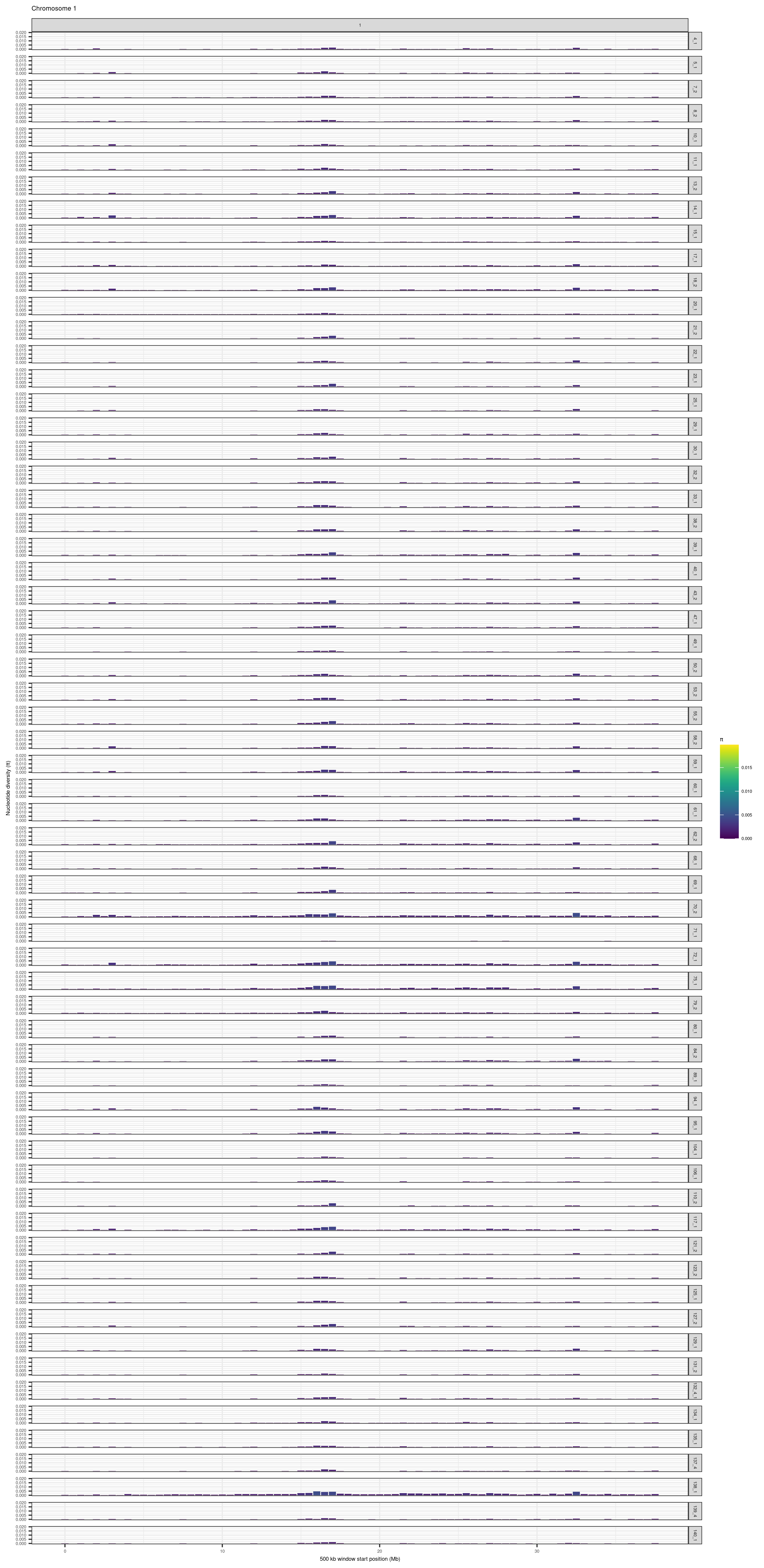
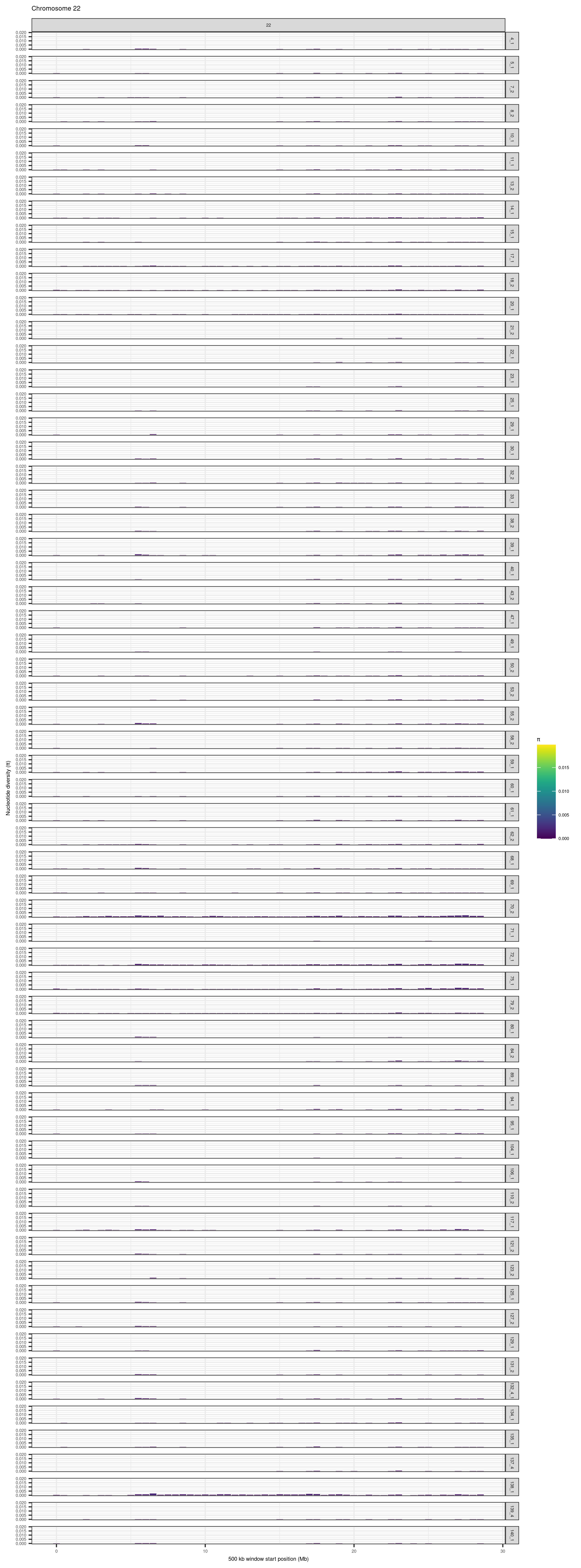
6.3.5 Boxplots
# source `darker` function
source("https://gist.githubusercontent.com/brettellebi/c5015ee666cdf8d9f7e25fa3c8063c99/raw/91e601f82da6c614b4983d8afc4ef399fa58ed4b/karyoploteR_lighter_darker.R")
# What is the median for MIKK v WILD?
pi_df_filt %>%
dplyr::group_by(POPULATION) %>%
dplyr::summarise(MEAN_PI = mean(PI, na.rm = T),
MEDIAN_PI = median(PI, na.rm = T))## # A tibble: 2 × 3
## POPULATION MEAN_PI MEDIAN_PI
## <chr> <dbl> <dbl>
## 1 MIKK 0.000298 0.000082
## 2 WILD 0.000253 0.00008# Together
mikk_wild_boxplot = pi_df_filt %>%
ggplot() +
geom_violin(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION)) +
geom_boxplot(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION), width = 0.1) +
theme_bw() +
scale_fill_manual(name = "Population", values = pal) +
scale_colour_manual(name = "Population", values = darker(pal, 80)) +
ylab(expression(paste(log[10], "(", pi, ")", sep = ""))) +
ggtitle("Nucleotide diversity in 500 kb bins (MAPQ >= 50 & GQ >= 40 & DP >= 40 )")
mikk_wild_boxplot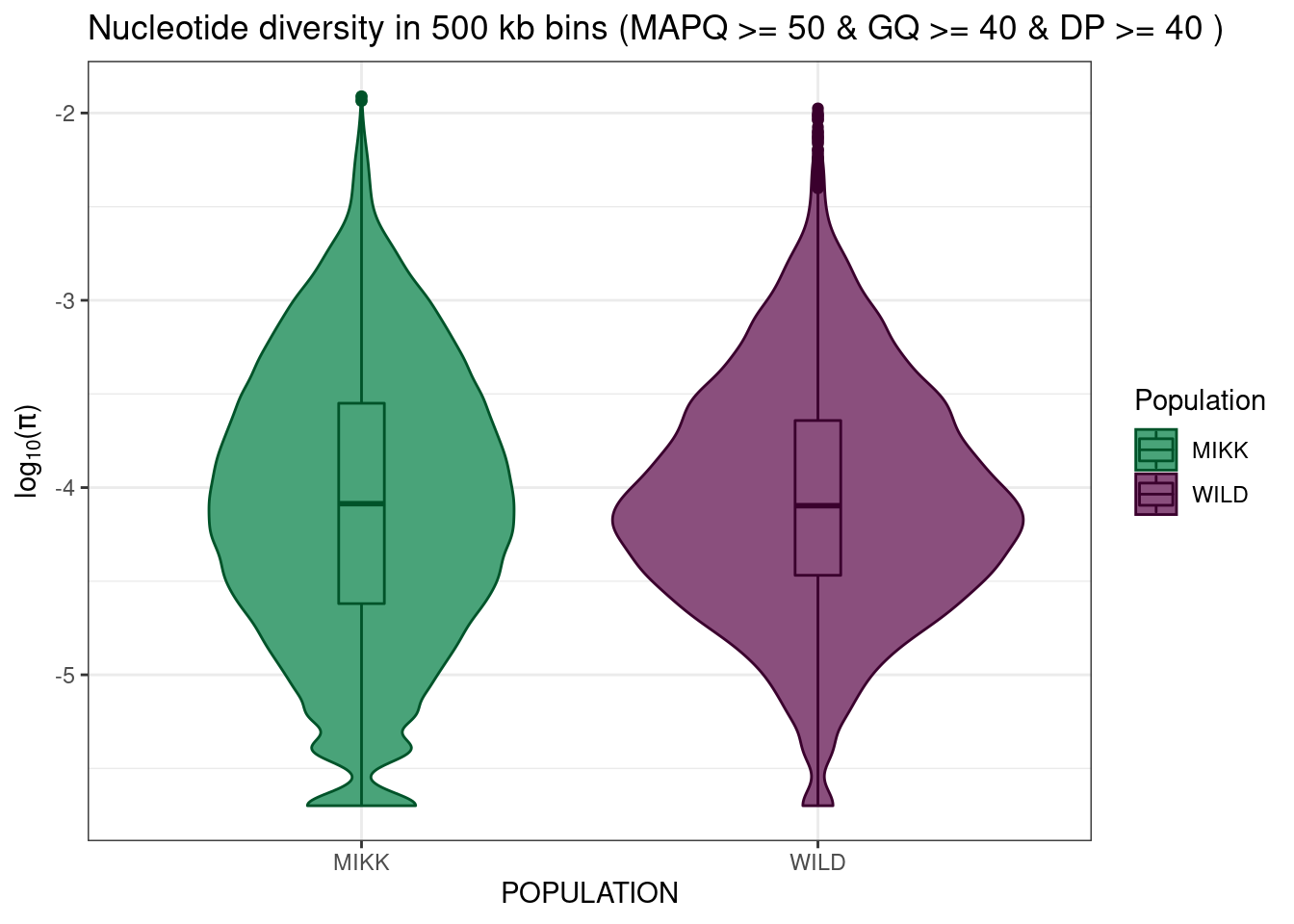
ggsave(here::here("docs/plots/nucleotide_diversity/20211006_mikk_wild_boxplot.png"),
plot = mikk_wild_boxplot,
device = "png",
width = 8,
height = 6,
units = "in",
dpi = 400)6.3.5.1 Take only 7 MIKK samples
set.seed(13)
# Pull out 7 random MIKK samples
mikk_random_samples = pi_df_filt %>%
dplyr::filter(POPULATION == "MIKK") %>%
dplyr::distinct(LINE) %>%
dplyr::slice_sample(n = 7) %>%
dplyr::pull(LINE)
# Plot
mikk_wild_boxplot_samp = pi_df_filt %>%
dplyr::filter(POPULATION == "WILD" | LINE %in% mikk_random_samples) %>%
ggplot() +
geom_violin(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION)) +
geom_boxplot(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION), width = 0.1) +
theme_bw() +
scale_fill_manual(name = "Population", values = pal) +
scale_colour_manual(name = "Population", values = darker(pal, 80)) +
ylab(expression(paste(log[10], "(", pi, ")", sep = ""))) +
ggtitle("Nucleotide diversity in 500 kb bins (MAPQ >= 50 & GQ >= 40 & DP >= 40 )\n7 MIKK lines & 7 wild Kiyosu samples")
mikk_wild_boxplot_samp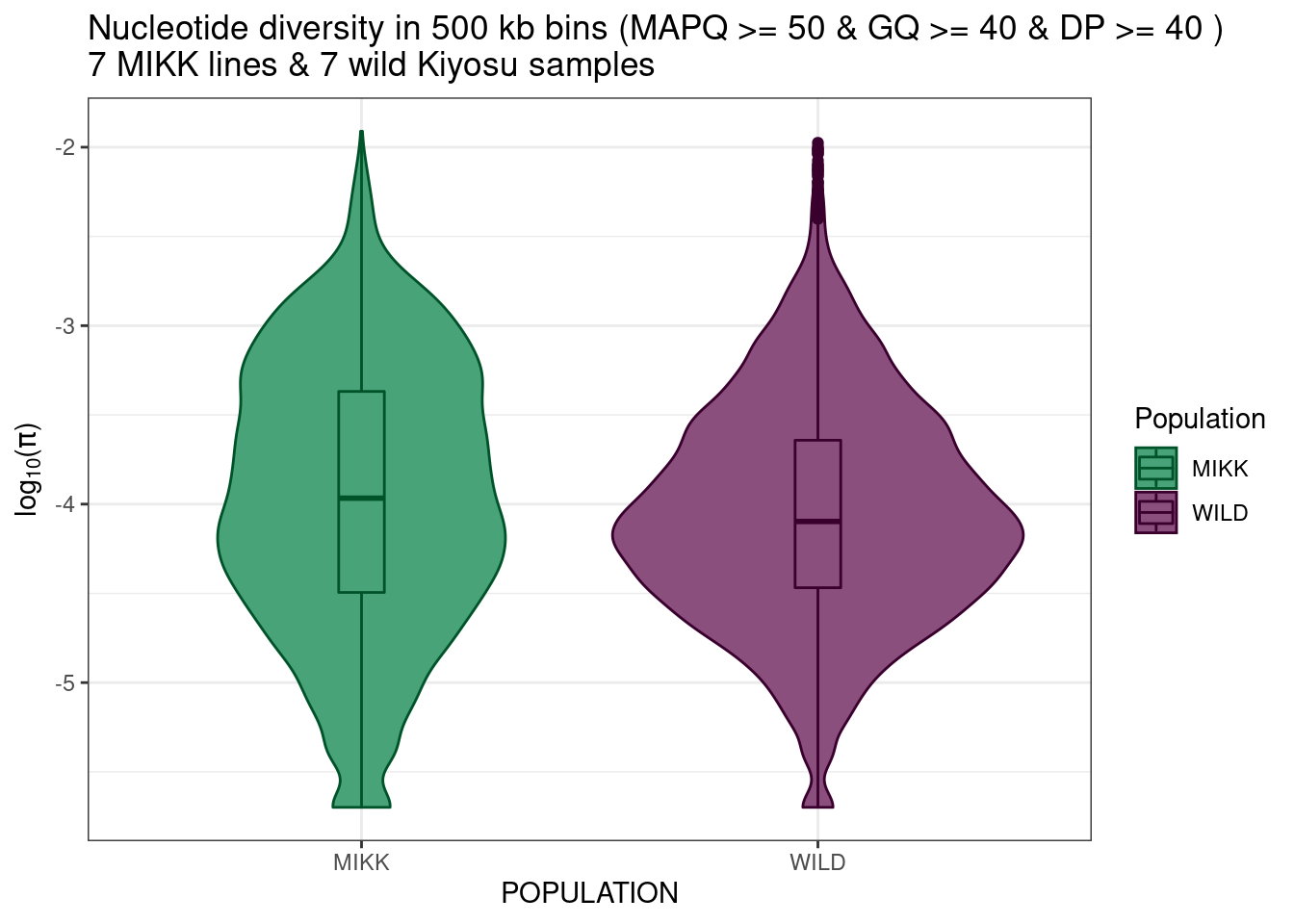
ggsave(here::here("docs/plots/nucleotide_diversity/20211011_mikk_wild_boxplot_sampled.png"),
plot = mikk_wild_boxplot_samp,
device = "png",
width = 8,
height = 6,
units = "in",
dpi = 400)6.3.5.2 Faceted
mikk_wild_boxplot_facet = pi_df_filt %>%
ggplot() +
geom_violin(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION)) +
geom_boxplot(aes(POPULATION, log10(PI), colour = POPULATION, fill = POPULATION), width = 0.05) +
theme_bw() +
scale_fill_manual(name = "Population", values = pal) +
scale_colour_manual(name = "Population", values = darker(pal, 80)) +
ylab(expression(paste(log[10], "(", pi, ")", sep = ""))) +
facet_wrap(vars(CHROM), nrow = 4, ncol = 6) +
guides(colour = "none", fill = "none") +
ggtitle("Nucleotide diversity in 500 kb bins (MAPQ >= 50 & GQ >= 40 & DP >= 40 )")
mikk_wild_boxplot_facet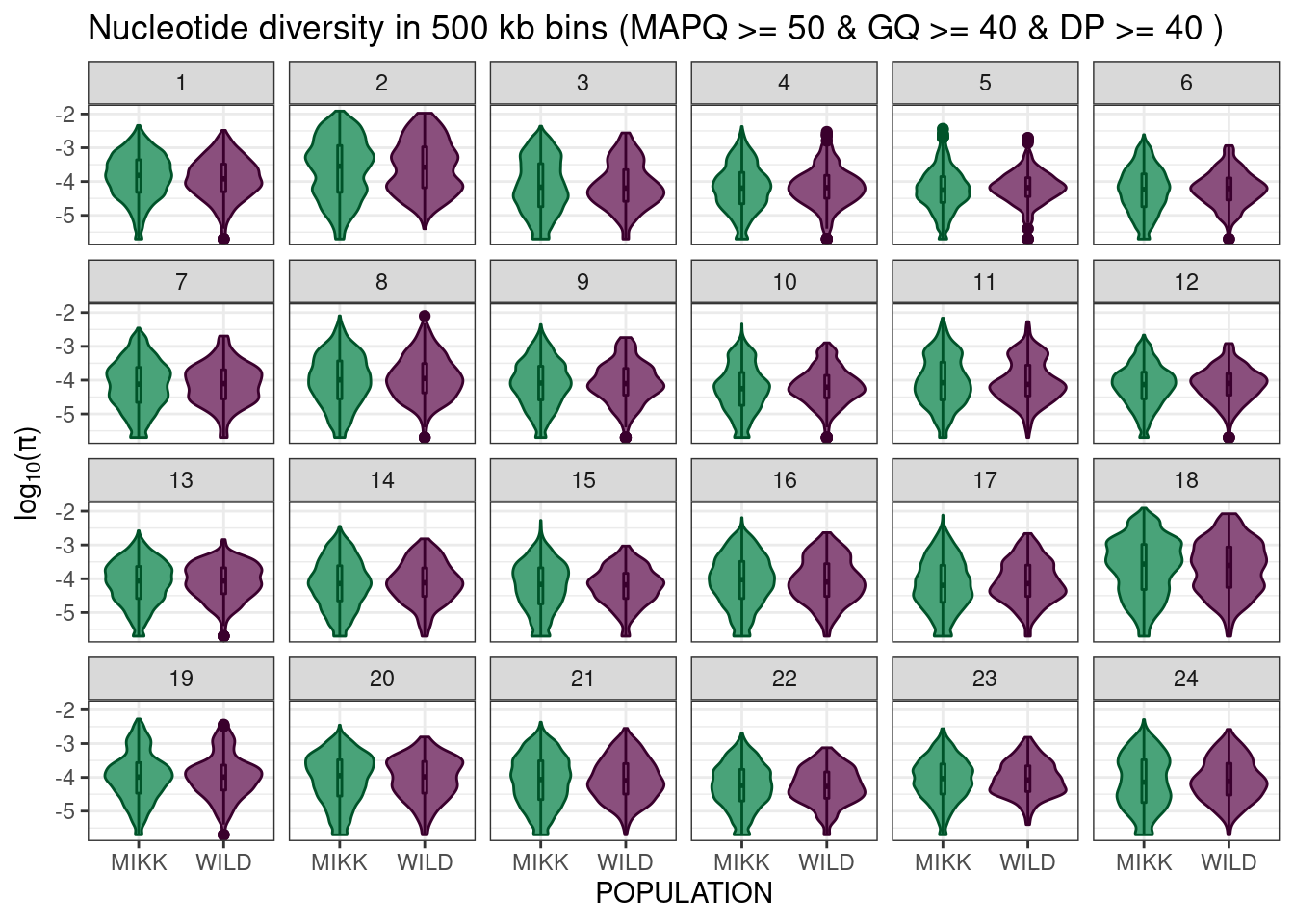
ggsave(here::here("docs/plots/nucleotide_diversity/20211006_mikk_wild_boxplot_facet.png"),
plot = mikk_wild_boxplot_facet,
device = "png",
width = 8,
height = 6,
units = "in",
dpi = 400)