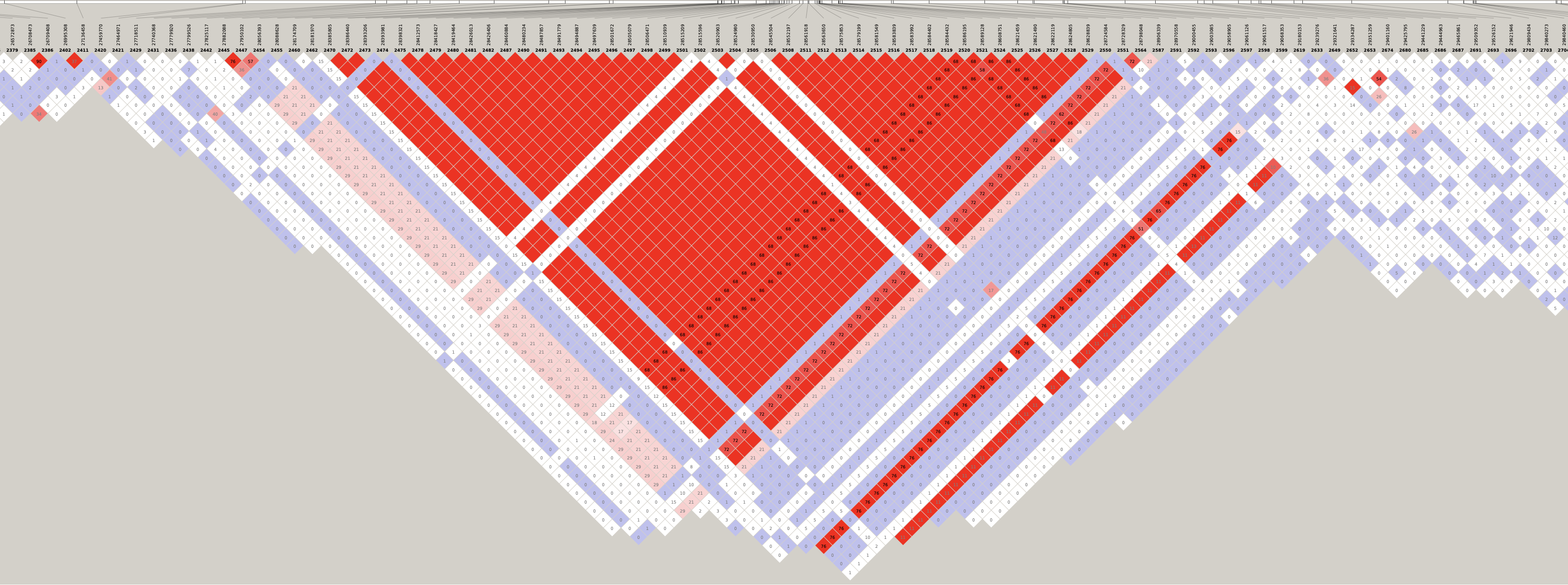
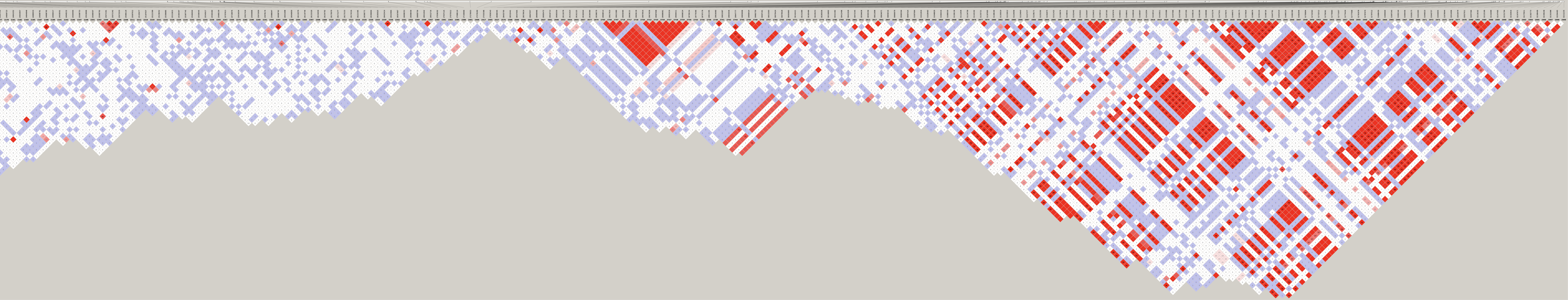
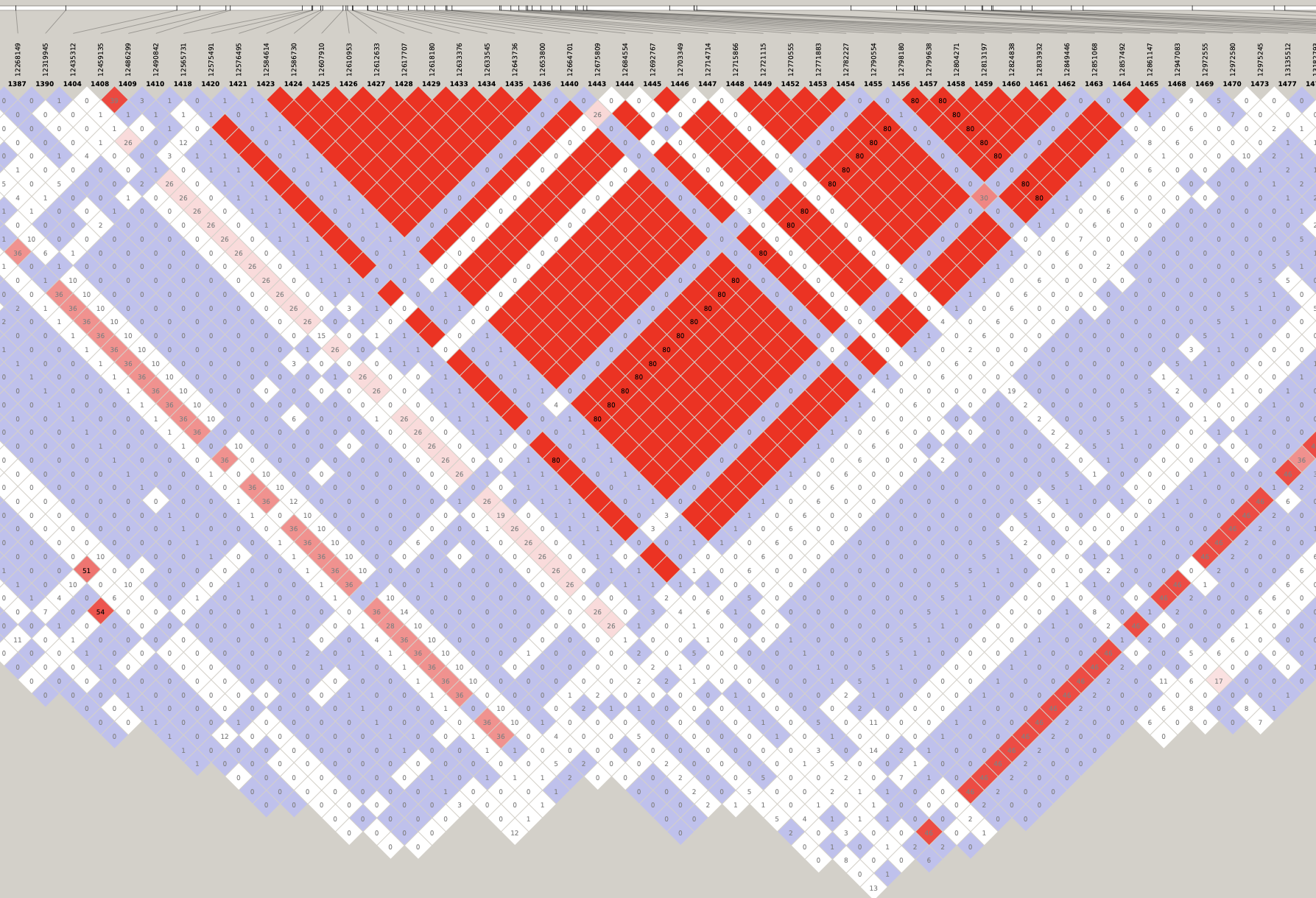
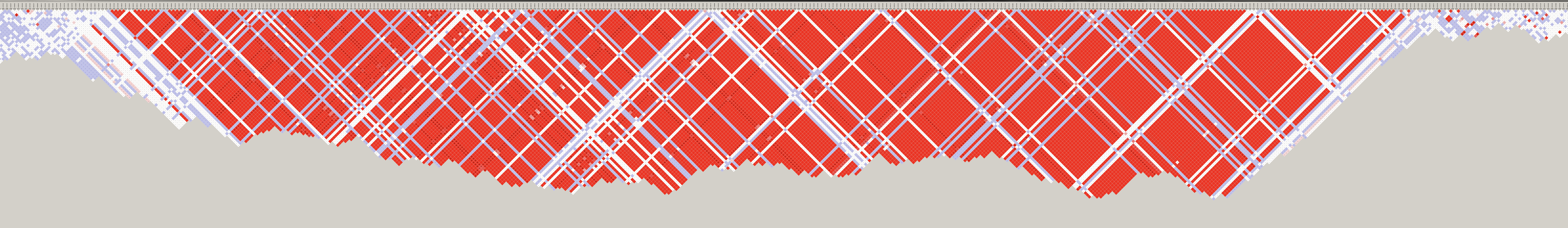
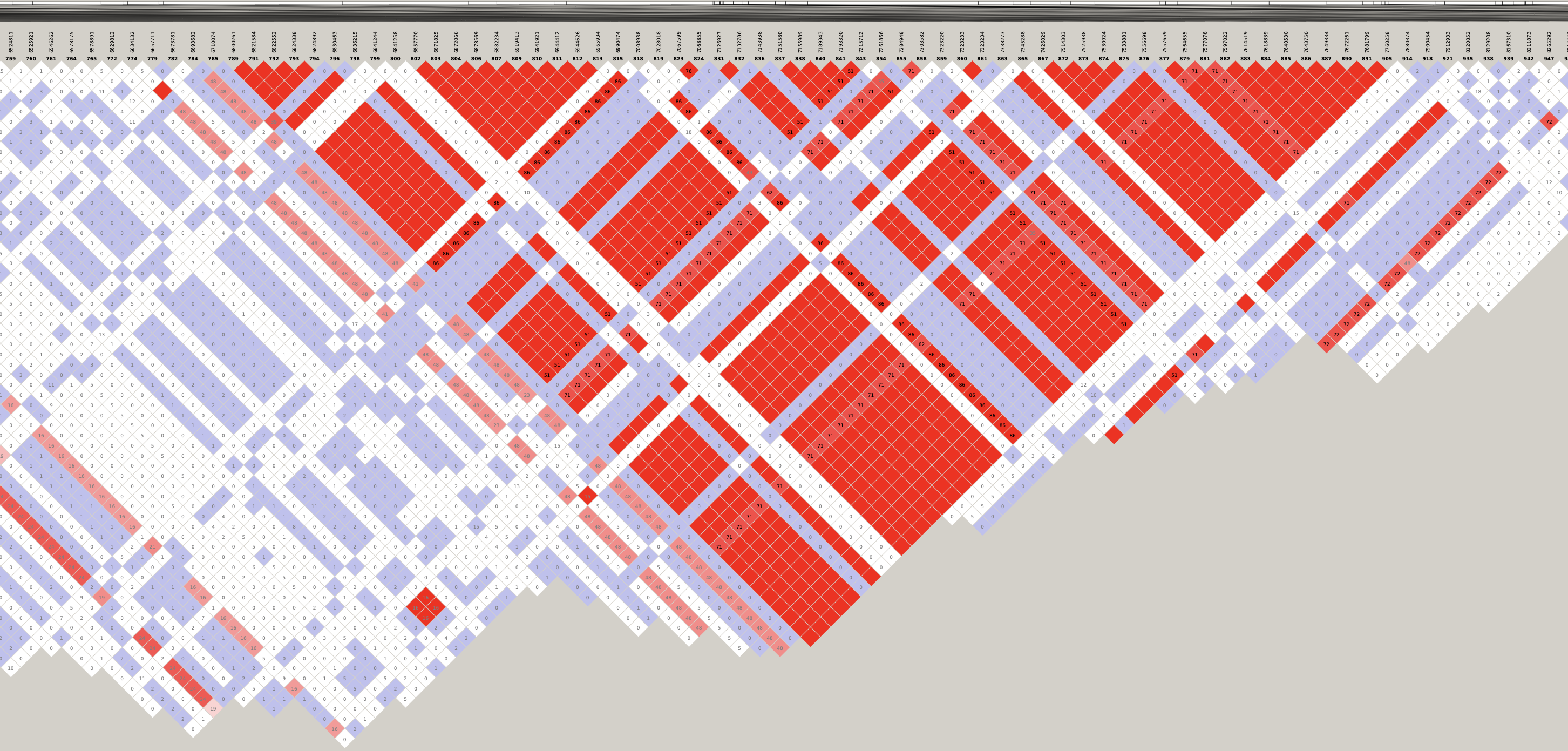
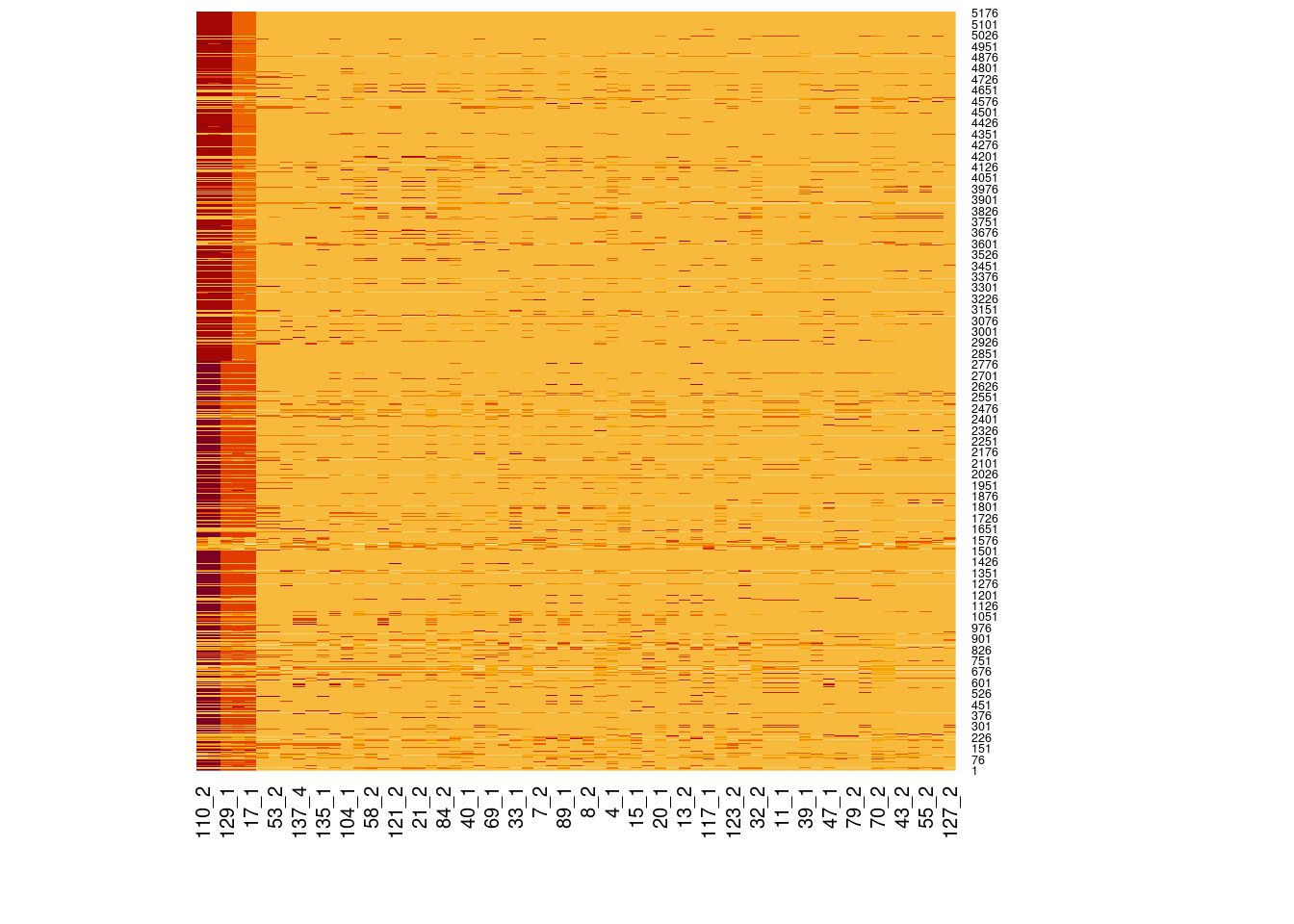
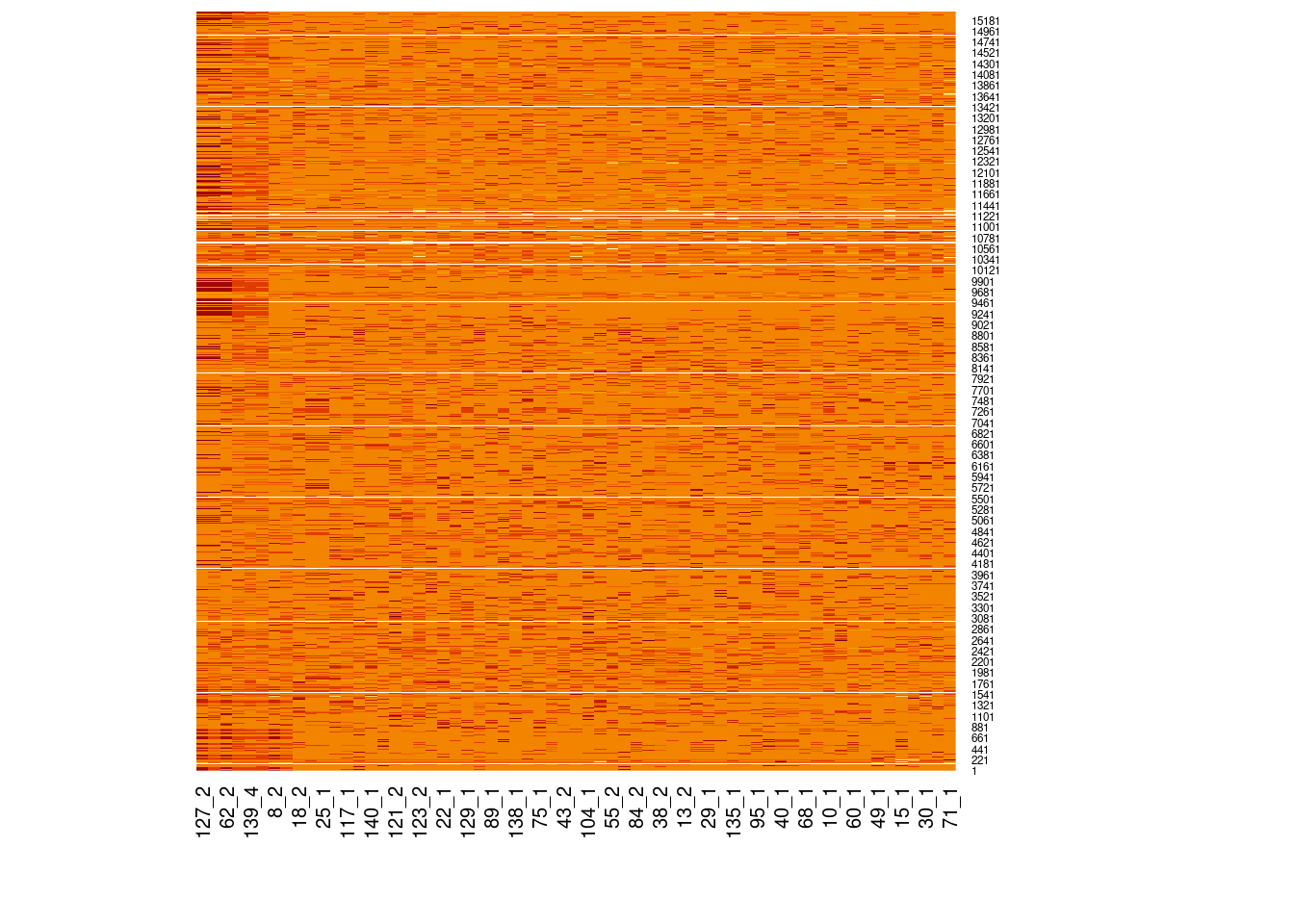
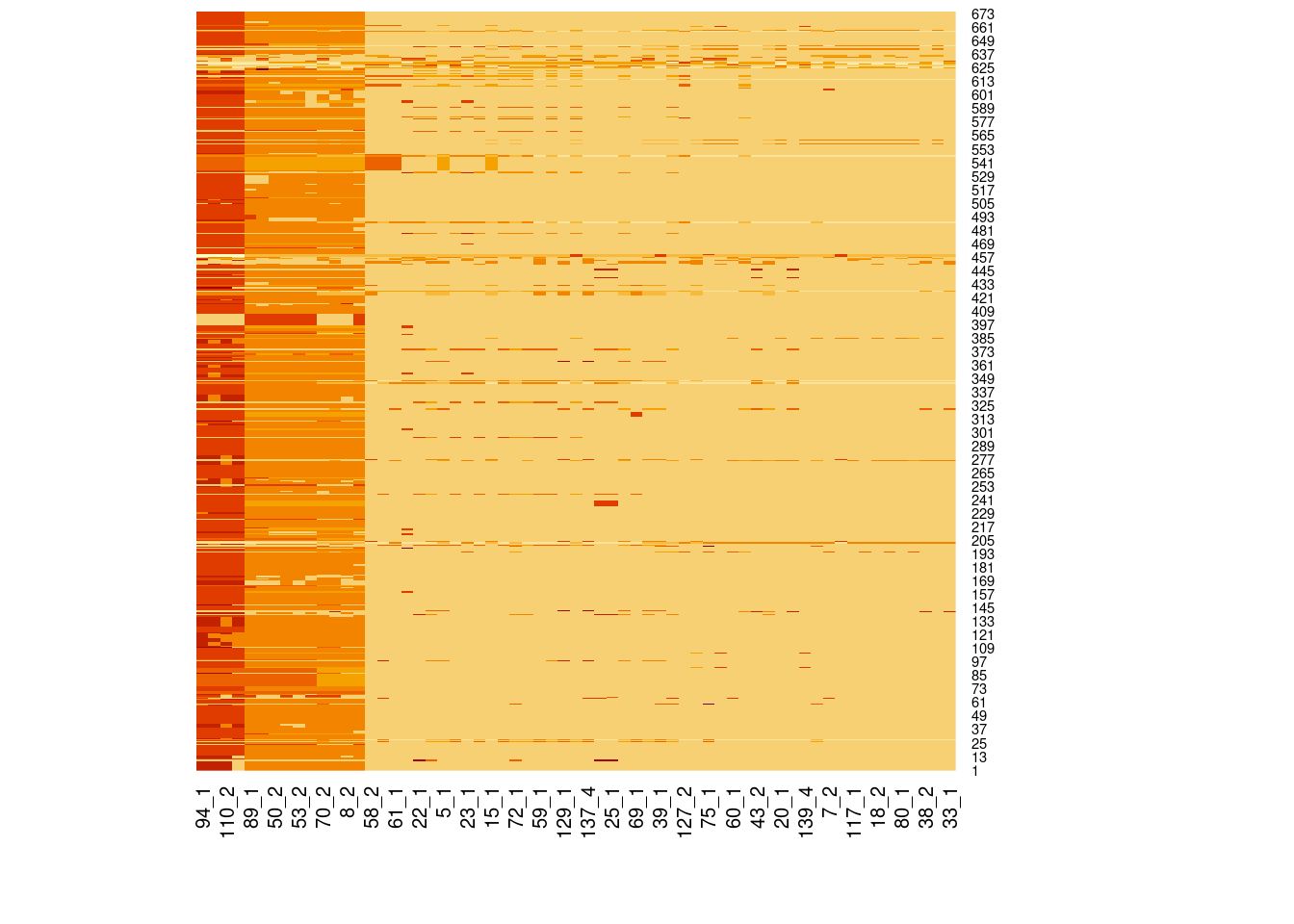
# Load libraries
library(here)
source(here::here("code", "scripts", "ld_decay", "source.R"))
# Get length of chromosomes
chr_counts <- readr::read_tsv(here::here("data",
"Oryzias_latipes.ASM223467v1.dna.toplevel.fa_chr_counts.txt"),
col_names = c("chr", "length")) %>%
dplyr::filter(chr != "MT") %>%
dplyr::mutate(chr = as.integer(chr))
# Select database and list datasets within
ensembl_mart <- useMart("ENSEMBL_MART_ENSEMBL")
# Select dataset
ensembl_olat <- useDataset("olatipes_gene_ensembl", mart = ensembl_mart)
olat_mart = useEnsembl(biomart = "ensembl", dataset = "olatipes_gene_ensembl")
# Get attributes of interest (exon ID, chr, start, end)
exons <- getBM(attributes = c("chromosome_name",
"ensembl_gene_id",
"ensembl_transcript_id",
"transcript_start",
"transcript_end",
"transcript_length",
"ensembl_exon_id",
"rank",
"strand",
"exon_chrom_start",
"exon_chrom_end",
"cds_start",
"cds_end"),
mart = olat_mart)
# Factorise chr so it's in the right order
chrs <- unique(exons$chromosome_name)
auto_range <- range(as.integer(chrs), na.rm = T)
non_auto <- chrs[is.na(as.integer(chrs))]
chr_order <- c(seq(auto_range[1], auto_range[2]), non_auto)
exons$chromosome_name <- factor(exons$chromosome_name, levels = chr_order)
# Convert into list
exons_lst <- split(exons, f = exons$chromosome_name)
# Get mean length of exons per chromosome
exons_lst <- lapply(exons_lst, function(chr){
chr <- chr %>%
dplyr::mutate(exon_length = (exon_chrom_end - exon_chrom_start) + 1,
transcript_total_length = (transcript_end - transcript_start) + 1)
return(chr)
})
# Get total length of chr covered by exons
exon_lengths <- lapply(exons_lst, function(chr){
# create list of start pos to end pos sequences for each exon
out_list <- apply(chr, 1, function(exon) {
seq(exon[["exon_chrom_start"]], exon[["exon_chrom_end"]])
})
# combine list of vectors into single vector and get only unique numbers
out_vec <- unique(unlist(out_list))
# get length of out_vec and put it into data frame
out_final <- data.frame("exon_cov" = length(out_vec))
return(out_final)
})
# combine into single DF
exons_len_df <- dplyr::bind_rows(exon_lengths, .id = "chr") %>%
dplyr::mutate(chr = as.integer(chr))
# join with chr_counts and get proportion of chr covered by exons
chr_stats <- dplyr::left_join(chr_counts, exons_len_df, by = "chr") %>%
dplyr::mutate(prop_cov_exon = exon_cov / length)
# convert chr to factor for plotting
chr_stats$chr <- factor(chr_stats$chr)